Answers to Questions about Analytical Method Validation, Verification, and Transfer
Reliable analytical results help in making informed decisions about the quality and safety of the products in the pharmaceutical industry. Also, such analytical data are essential to support the drug product registrations. Learning how to execute document development, how to demonstrate FDA and EU compliance to auditors and inspectors, how to explain company's strategy for method validation, verification, transfer and equivalency testing and the best practices will be invaluable to your firm.

This article provides answers questions about Analytical Method Validation, Verification and Transfer and guides you to the next steps. Scientists who are directly or indirectly involved with the drug development, analysis, stability studies or regulatory/compendial submissions will find the information in this article useful. Among others who will benefit from this article are Analytical Chemists, supervisors and managers of Analytical Chemists, those responsible for hosting FDA inspections, responding to 483s and warning letters, regulatory affairs or those preparing for NDA's and ANDAs, QA managers, Quality control staff, validation specialists, consultants, formulation chemists, lab supervisors, Compendial Liaisons, and chemistry, pharmaceutical, pharmacy senior or graduate students.
What is method validation?
Why is method validation necessary?
- Good science
- Regulatory requirement
- An Essential part of Good manufacturing practice (GMP)
- In practice, it is usually possible to design the experimental work such that the appropriate validation characteristics can be considered simultaneously, to provide a sound, overall knowledge of the capabilities of the analytical procedure, for instance; Specificity, Linearity, Range, Accuracy, and Precision.
- Support the identity, strength, quality, purity, and potency of the drug substances and drug products.
- Validation of Analytical Procedures: Text and Methodology Q2(R1), ICH 2005
- Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics, FDA 2015
- Guidance for Industry: Bioanalytical Method Validation (Draft Guidance), FDA 2013
- <1225> Validation of Compendial Procedures, USP 39
- <1226> Verification of Compendial Procedures
- <1058> Analytical Instrument Qualification.
What is the lifecycle of an analytical lifecycle?
Where is the Analytical validation performed?
Who should perform the analytical validation?
When should the Analytical validation be performed?
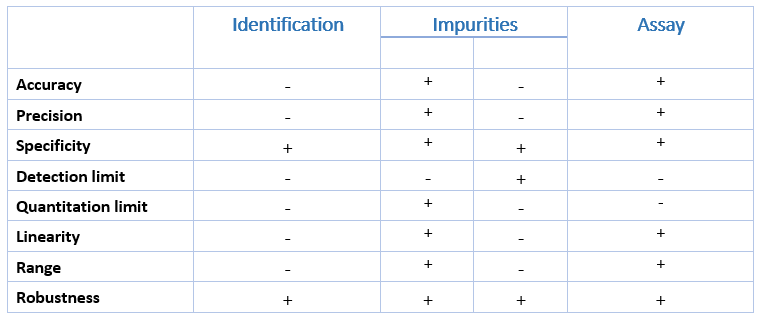
What Analytical Procedures should be validated?
- Identification tests
- Quantitative tests for impurities content
- Limit test for control of impurities
- Quantitative tests for the active moiety of the drug substance(s), drug product or other selected components in the drug
- Dissolution testing and determination of particle size
When is the analytical method validation required?
- Prior to the use of the method in ongoing or routine testing
- When there are changes to previously-validated conditions or method parameters, if changes are over the scope intended by the original method
- When the method is part of a New Drug Application (NDA) or Abbreviated New Drug Application (ANDA) submission
What are the required validation documents?
- Protocol - Procedures and acceptance criteria
- Report - Documented results
- Justification - when non-pharmacopoeial methods are employed
- Detailed standard test methods
What characteristics should be considered during the validation of analytical methods?
- Specificity
- Linearity
- Range
- Accuracy
- Precision
- Detection limit
- Quantitation limit
- Robustness
How should analytical validation be performed?
How is the degree of validation determined?
What are the other integral parts of analytical method validation?
- The Resolution, tailing factor, RRT, capacity factor, number of theoretical plates between two peaks in chromatography methods.
- Spike recovery in sample processing is present
- Minimum height /response of control samples
- BC recovery of the known amount of control sample
- Every parameter should comply or pass. Failure will be considered as not used.
What is method verification?
When should verification or revalidation be done?
- When there are changes in the process for the synthesis of the drug substance.
- When there are changes in the composition of the finished product.
- When there are changes in the analytical procedure
- When there is a transfer of methods from one laboratory to another.
- When there are changes in major pieces of equipment instruments.
- The Degree of validation depends on the type of the change(s)/ its criticality
What is 'Transfer of Analytical Method'?
Why is Controlled Method Transfer necessary?
- It is a Regulatory requirement.
- 'The suitability of all testing methods used shall be verified under actual condition of use'
- 21 CFR Part 211.194 (a) (2) - 'Laboratory records shall include a statement of each method used in the testing of the sample.'
- 'The statement shall indicate the location of data that establish that the methods used in the testing of the sample meet proper standards of accuracy and reliability as applied to the product tested'
- 'The suitability of all testing methods used shall be verified under actual condition of use.'
- Controls failure rate
- Improves the quality of the testing at the new lab
What are the documentation requirements for Method transfer?
- Transfer Master Plan (TMP) used as framework
- Transfer project plan or method transfer protocol (MTP)
- Approach for controlled transfer and justification
- SOPS for step-by-step implementation
- Forms for efficiency and consistency
- Analytical procedure
- Original validation report
- Historical data on method reliability and performance (to identify the biggest reasons for variance)
What should the Method Transfer report consist of?
- Summary of method familiarization results
- Detailed results of the transfer study
- Description of any deviations and from expected results and how they have been resolved
- Signatures of individuals responsible for transfer studies
- Copies of supporting material
- Conclusion and assessment of the transfer study results
'The confirmation by examination and the provision of objective evidence that the particular requirements for a specific intended use are fulfilled.' - ISO/IEC 17025 definition.
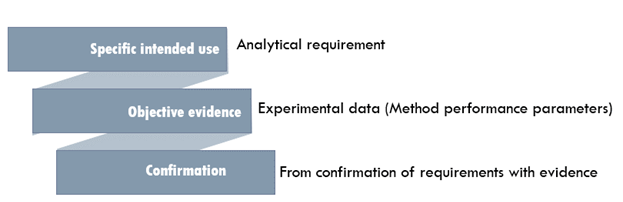
More definitions
"Establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes" - US FDA 1987 definition "Collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality products." - US FDA 2011 definition "Action of proving, in accordance with the principles of GMP, that any procedure, process, equipment, material, activity or system actually leads to the expected results." - World Health Organization (WHO)Good science
Method validation establishes the fitness-for-purpose on the customer's behalf. Conducting method validation is also good science.
Regulatory requirements
Analytical data are required for regulatory submissions in support of the drug product registrations. The Regulatory bodies require proof of safety and quality of products. Companies are required to have reliable test methods. The reliability of test methods is substantiated by performing validation which is then documented.
The Code of Federal Regulations (CFR) 311.165c explicitly states that the "Accuracy, Sensitivity, Specificity, and Reproducibility of test methods employed by the firm shall be established and documented.
ICH GUIDELINE Q2 (R1)
The objective of validation of an analytical procedure is to demonstrate that it is suitable for its intended purpose,
Guidance Documents
Validation is an essential part of GMP practice.
Reliable analytical results are necessary to make informed decisions about the quality and safety of the products in the pharmaceutical industry.
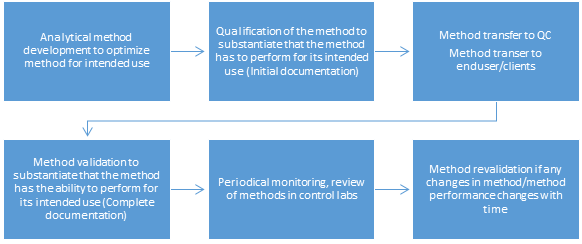
Analytical validation is usually performed in a GxP laboratory or equivalent. Ideally, it should be conducted in the end-user (QC) laboratory where the method is to be employed regularly.
It is also important to use certified reference material in analytical laboratories to meet the FDA and international requirements for selection, purchasing, testing, storage, and use of (certified) reference material.
GxP trained, skilled and qualified analysts should perform the analytical validation.
Analytical validation should be performed before the commercialization
Results should be accurate, reliable, and reproducible.
The regulatory bodies such as have provided many guidelines on how to perform analytical validation. These include ICH Q2, FDA, USP, EMEA.
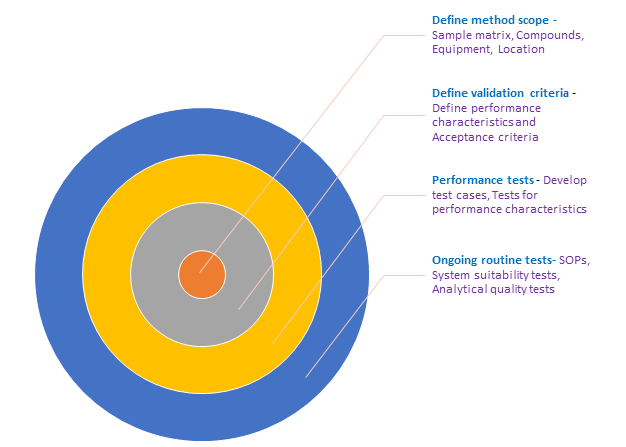
The degree of validation depends on the test.
System suitability and acceptance criteria are integral parts of analytical validation. During each method/procedure, it should be ensured that all analytical operations, electronics and equipment are working properly at the time of analysis. Relevant system suitability, and its criteria should be defined including
Method verification is the provision of objective evidence that a given item fulfills specified requirements (ISO/IEC Guide 99:2007)
Verification USP <1226>
The Analytical procedures in the current USP are legally recognized under section 501(b) of the Federal Food, Drug and Cosmetic Act as the regulatory analytical procedures for the compendial items. The suitability of these procedures must be verified under the actual conditions of use.
When using USP analytical procedures, the guidance recommends that information is provided for the following characteristics:

Non-pharmacopoeial methods appropriately validated.
USP <1224> 'The transfer of analytical procedures (TAP), also referred to as method transfer, is the documented process that qualifies a laboratory (the receiving unit) to use an analytical test procedure that originated in another laboratory (the transferring unit), thus ensuring that the receiving unit has the procedural knowledge and ability to perform the transferred analytical procedure as intended."
You can get a clearer understanding of the Transfer of Analytical methods according to USP Chapter <1224> here.
A drug's characterization, quality control, and manufacturing batch records are supported by validated analytical records. Analytical method validation provides documented proof that the test procedure is suitable for its intended. It provides evidence of the method's performance and the quality and reliability of results. Analytical methods provide data that is important to ensure consumer health and safety. Hence their validation is highly inspected.
A drug's characterization, quality control and manufacturing batch records are supported by validated analytical records. To show that the test procedure is suitable for its intended analytical test method is documented, thus providing proof of the method's performance and quality and reliability of results.
Attend the seminar Analytical Method Validation, Verification and Transfer to understand how to determine the analytical characteristics for different types of validation procedures for the analysis of both the drug substance and drug product. The factors to consider for verification of the compendial procedures will also be discussed in the seminar. In addition, different approaches for the transfer of analytical procedure from one lab (transferring) to other lab(s) (receiving) under different circumstances will be covered. Other related topics for obtaining reliable data will also be discussed. These topics include analytical instrument qualification as well as how to set, handle and monitor specifications.
The course Director Ms. Thomas has over two decades of cGMP hands-on industry experience in both pharmaceutical and medical device manufacturing operations. Her experience covers all Quality Systems; as well as, all areas of validation; including, process/product validation, facilities validation, CSV and 21 CFR Part 11, test method validation, equipment/automated processes and cleaning validation. Utilizing strategic thinking, risk-based approaches, and Lean principles, she has demonstrated success in steering and managing complex projects within the pharmaceutical and medical device industries.




