The Basics of Extractables or Leachables studies for Biologics - Definitions, Regulatory requirements, Guidelines, and Program Initiation
The FDA has increased scrutiny for extractable and leachable associated biotechnology drug products in recent years. Biologics products are particularly sensitive to leachable. Companies are increasingly looking for better ways to conserve the time and resources required for extractable or leachable studies through proper planning. However, prior to planning the extractable or leachable studies, it is important to understand its basics and the related regulatory and industry guidance. This article sets the stage.

The basics
Drugs and medical devices must be free of harmful materials before administering them to a patient. To ensure patient safety, the FDA requires all Drugs to be qualified to ensure that no chemicals transfer into the drug from its packaging or production. Such an analysis is called Extractables and Leachables Testing. Drug companies use these tests to ensure and demonstrate that the potentially harmful materials are not transferred from the packaging, production to the patient.
In short, the purpose of Extractables testing is:
- To qualify CCS (container/closure system) components
- To screen for and monitor if toxic materials are present
- To develop analytical methods for leachable testing
- To show quality control for acceptance of CCS components
And the purpose of Leachables testing is:
- To monitor and control impurities derived from CCS in the DP (Drug products) during stability and/or as part of container qualification studies
The FDA's definitions of Extractables and Leachables
- Extractables: Compounds that can be extracted from the container closure system when in the presence of a solvent
- Leachables: Compounds that leach into the drug product formulation from the container closure as a result of direct contact with the formulation
Regulatory and Industry Guidance for Extractables and Leachables testing
- 21CFR part 211.94: (a) Drug product containers and closures shall not be reactive, additive, or absorptive so as to alter the safety, identity, strength, quality, or purity of the drug beyond the official or established requirements.
- Container Closure Systems for Packaging. Human Drugs and Biologics. Chemistry Manufacturing, And Controls (FDA, May 1999)
- Metered Dose Inhaler and Dry Powder Inhaler Drug Products Chemistry, Manufacturing and Controls Documentation (FDA, Oct 1998)
- Industry Nasal Spray and Inhalation Solution Suspension, and Spray Drug Products Chemistry, Manufacturing and Controls Documentation (FDA, July 2002)
- Guideline on Pharmaceutical Quality of Inhalation and Nasal Products (EMEA, June 2006)
- Guideline on Plastic Immediate Packaging Materials (EMEA, May 2005)
Study requirements - Pre-NDA Division Boilerplate Comment
'The NDA submission should contain information on extractables and leachables from the drug container closure system and/or drug product formulation unless specifically waived by the Division.
Solvents and conditions used in extraction studies should be justified.
Results of extraction studies should be used to assure that drug product stability samples are adequately monitored for potential leachables.
A risk assessment (RA) based on results of extraction studies may be adequate to support safety during development, the RA of leachables in the drug product identified over the course of stability studies will form the basis for the final safety determination.
The approach for toxicological evaluation of the safety of leachables must be based on good scientific principles and take into account the specific container closure system, drug product formulation, dosage form, route of administration, and dose regimen (chronic or short-term dosing).
For your toxicological risk assessment, any leachable that contains a structural alert for mutagenicity should not exceed 1.5 mcg/day total daily exposure for a chronic indication or be adequately qualified for safety. From a genetic toxicology perspective, we will allow up to 120 mcg/day for an acute indication for most potentially genotoxic impurities.
A toxicological risk assessment should be provided for any nongenotoxic leachable that exceeds 5 mcg/day. The risk assessment should be based on the levels of leachables detected in long-term stability samples.
The safety assessment should be specifically discussed in Module 2.6.6.8 (Toxicology Written Summary/Other Toxicity) of the NDA submission.'
Sources of Leachables
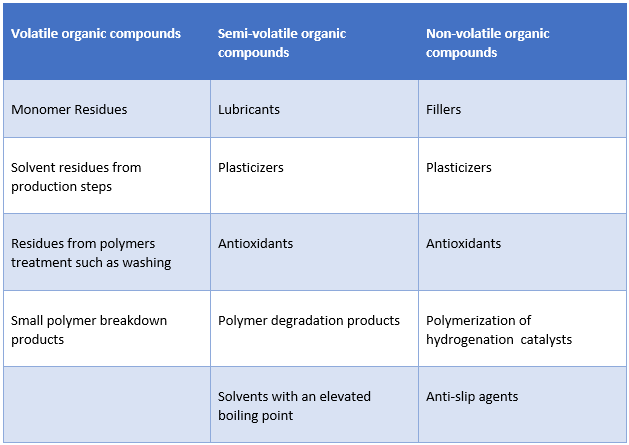
Control of supply chain to monitor for and prevent contamination
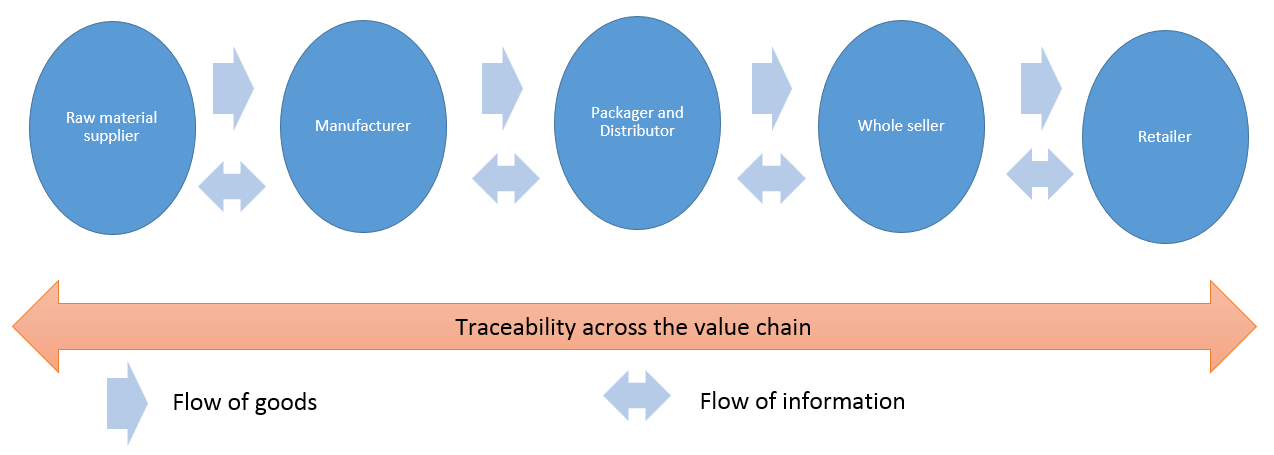
Transparency is required throughout the pharmaceutical supply chain. Starting from the suppliers, transparency must be maintained all the way down the supply chain.
Initiating an Extractables and Leachables program
- Understand the process
- Tubing and connectors that transfer starting buffers into a process stream
- Syringes and needles that transfer liquids
- Sterile and nonsterile filters that filter the starting buffers
- Bags and tubing that store and transfer intermediate process formulations
- Tangential-flow (cross-flow) filters used for concentration and diafiltration of process intermediates
- Column chromatography sorbents and chromatography membranes used for concentration and purification of process intermediates
- O-rings used to seal connectors and sanitary fittings.
- Final bulk containers (including closure materials of construction) and filters used before final filling.
- Perform risk assessment
- Materials compatibility
- The proximity of components to a final product
- The composition of the product
- The surface area of the product-contact components
- Contact time and temperature
- Pretreatment steps
Understand your manufacturing process from the beginning to the end. Derive a detailed manufacturing process using batch records and associated standard operating procedures (SOP), technical reports, and batch analyses. Resolve any discrepancies between those documents to ensure you represent the process accurately. Define all the materials that have product contact including
The above is not an exhaustive list. There is no need for analyzing further if there is no product contact as given above
Risk factors include
Once the above two steps are performed, you can get into the detailed Extractables and Leachables study design.
Attend the webinar 'Extractables and Leachables in Early Phase Development' to understand the steps of an extractables / leachables study, get knowledge about the gap analysis of literature sources and how to design an efficient E/L study.
The instructor Dr. Subbarao, has hands-on industrial experience covers stability and laboratory cGMP systems for both biologics and conventional drugs. She has extensive experience in evaluation of analytical methods and method validation for products ranging from conventional drugs, well- characterized proteins, vaccines, cell therapy and gene therapy products, ranging from pre-clinical phase to commercial phases. She holds a Ph.D. in Bio-organic Chemistry from the Indian Institute of Technology, Bombay, India.




