cGMP and GLP Regulations and Inspections for Quality Control Labs - An overview
A large number of quality control related 483s and warning letters in recent years demonstrate that regulated companies are having problems with the implementation of regulations for quality control laboratories. FDA and other agencies have held inspections of QC laboratories in the highest emphasis because the drug products and APIs (Active Pharmaceutical Ingredient) are released to the market after testing and approval without any further check. This article provides the regulatory background and informs you of what the inspectors will check during inspections.
In addition to reading this article, a demonstration of how quality control and quality assurance personnel can monitor industry practices to stay "current" with FDA requirements (cGMPs and GLPs) will be helpful. The seminar Quality Control Laboratory Compliance - cGMPs and GLPs takes a deep dive into the topic.

GMP requirements for Quality control laboratories B'211.160
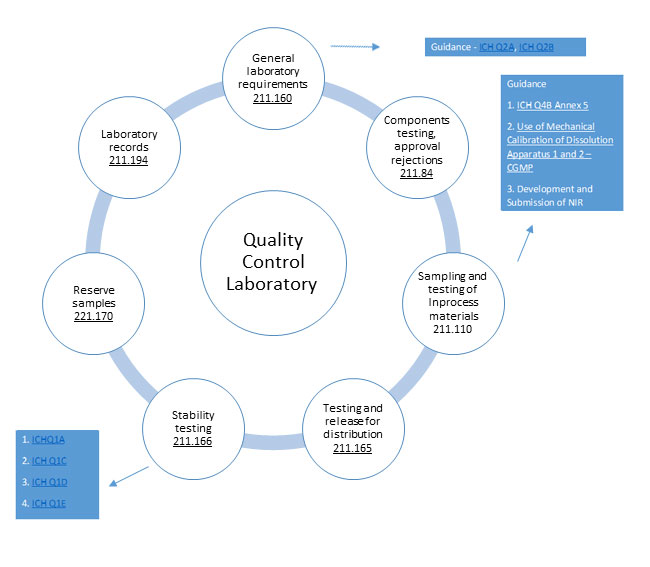
'General requirements:
- Scientifically sound and appropriate specs
- Establish written specs, sampling, procedures
- Conformity to written specifications
- Calibration of instruments
- Conformance to appropriate standards of identity, strength, quality, & purity
- Test results documented at the time of performance
- Appropriate procedures for the acceptance of components, in-process materials, and drug products
- Methods must be documented and approved
Testing and Approval or Rejection of Components:
- Hold back from use each lot of component, drug product containers, and closures until the lot has been sampled, tested, or examined and released for use
- The Representative sample consists of a number of units that are drawn based on rational criteria such as random sampling and intended to assure that the sample accurately portrays the material being sampled.
- At least one specific identity test is conducted on the component by the manufacturer at the time of receipt.
- The manufacturer establishes the reliability of the supplier's analysis by validating the supplier's test results at appropriate intervals.
Sampling and Testing of In-process Materials and Drug Products:
- Written procedures shall be established and followed for inprocess testing
- In-process materials shall be tested for identity, strength, quality, and purity, and approved or rejected by the QU
- Testing of samples shall assure that the drug product and in process material conform to specifications
Testing and Release for Distribution:
- Documented accuracy, sensitivity, specificity, and reproducibility of test methods
- Test each batch for conformance to specifications
- Follow written sampling and testing plans
- Document testing methods and number of units/batch
Drug Product Stability Testing:
- Written stability testing program
- Full shelf-life studies for determining the expiration date
- Combination of accelerated studies and basic stability
- Conducted on samples from representative batches
- Sample size and test intervals based on statistical criteria
Reserve samples:
- Reserve samples from representative sample lots or batches
- Stored under conditions consistent with product labeling
- Results of the examination shall be recorded and maintained with other stability data on the drug product
- Investigate any evidence of reserve sample deterioration
Laboratory Records:
- A description of the sample
- The location of the method validation data
- A description of the suitability of the method
- Record of the sample weight including all test data obtained
- A statement that test methods are accurate, reliable, and followed'
GLP regulations (21CFR part 58)
GLP regulations are published is CFR part 58 and apply to
Non-clinical laboratory studies that support or are intended to support application for research or marketing permits for the following products:
- food and color additives
- human and animal drugs
- medical devices for human use
- biological products
- electronic products
(58.3)
"In vitro or in vivo experiments in which test articles are studied prospectively in test systems under laboratory conditions to determine their safety. The term does not include studies utilizing human subjects or clinical studies or field trials in animals.
The term does not include basic exploratory studies carried out to determine whether a test article has any potential utility or to determine the physical or chemical characteristics of a test article."
Elements of GLP

Management Responsibilities (58.31):
- Before initiating the study, designate a Study Director
- Ensure there is a Quality Assurance Unit
- Ensure that the test and control article have been appropriately tested for identity, strength, purity, stability and uniformity, as applicable
- Authorize significant changes in SOPs
- Ensure that any deviation from GLP regulations reported by the quality assurance unit are communicated to the Study Director and corrective actions are taken and documented
Study Director:
"For each non-clinical laboratory study, a scientist or other professional of appropriate education, training and experience, or combination thereof, shall be identified as Study Director".
Quality assurance unit:
"A testing facility shall have a Quality Assurance unit which shall be responsible for monitoring each study to assure management that the facilities, equipment, personnel, methods, practices, records, and controls are in conformance with the regulations.
For any given study, the Quality Assurance unit shall be entirely separate from and independent of the personnel engaged in the direction and conduct of the study."
Testing Facility:
"A testing facility shall have standard operating procedures in writing setting forth non-clinical laboratory study methods that Management is satisfied are adequate to insure the quality and integrity of the data generated in the course of a study.
"Each laboratory area shall have immediately available laboratory manuals and standard operating procedures relative to the laboratory procedures being performed. Published literature may be used as a supplement to standard operating procedures."
"A historical file of standard operating procedures, and all revisions thereof, including the dates of such revisions, shall be maintained."
What the Inspectors will check during pharmaceutical quality control laboratory Inspections
General:
- General information - The contact person (name, phone No, email)
- Test activities - Whether licensed by a competent national authority, whether Regularly inspected by a competent national authority
- Contract testing - Whether licensed by a competent national authority, Evaluation / Re-evaluation of the contract laboratory by the customer (contract giver)
General:
- General information - Whether document is available, are the key personnel, reporting lines, whether responsibilities and release criteria
- Ensuring suppliers quality - Whether there is a clearly defined quality assessment policy, whether audits, qualification/evaluation made.
- Self-inspection - How and by whom was it performed? How is it reported? How are corrective measures implemented? Is the schedule available and adhered to?
- Trending - Assess trends, whether SOP exists, who and how are the trends evaluated?
- Change control - How are the changes documented, controlled and managed?
- Risk management - Risk management method/approach
Documentation:
General information - System description whether defined in writing, (format, numbering system, approval criteria, distribution, return, interval for revision etc.), what is the archiving process and how is it managed, whether SOP consists the authorization for copying, identification of copies from official and controlled documents
Laboratory documentation
- Specifications (SPECs), Do SOPs for sampling, testing, equipment handling and other laboratory processes exist, are they complete, adequate? The archiving of previous SOPs
- Test instructions, analytical procedures, methods
- Test records/test batch protocols
- Log books
- Raw data /e.g. chromatograms, spectra, results), out prints
Data traceability
- Procedure, record on receipt and usage of materials, standards
- Sample tracking
- Analytical raw data traceability
Electronic documentation/ computerized systems
- Same requirements whether paper or electronic
Personnel
- General - Number of employees (total), specified to positions and different testing
- QC Manager and deputy (other key personnel)
- Training - Training system, program, documentation, evaluation of the effectiveness of the training
Facility and equipment
- Facility - Location of the QC laboratories, Facility design, rooms separation, Temperature, humidity, ventilation and recording systems/alarms, Storage areas, labelling
- Equipment- Instrumentation, Assembly, Calibration, Labelling, Log books.
- Equipment validation: Qualification, Design qualification, Installation qualifications, Operational qualifications, Performance qualifications
- Cleaning - system of cleaning and sanitization, validation, documentation, time limits, equipment used cleaning. Cleaning personnel information, SOPs
- Maintenance - System, preventive maintenance, SOPs, documentation
Materials and supplies
- Materials - Laboratory reagents, standards, Reference substances, Handling of highly toxic, hazardous and sensitizing materials, poisons
- Water and water systems -water system quality, sampling, testing
Sampling and samples
- Sampling - General policy, sampling, Place of sampling for raw materials, Starting/packaging materials sampling, Intermediates sampling, air sampling system, water sampling system, procedures, records, retained samples, re-sampling
- Samples - Handling, retained samples, samples tracking
- Personnel for sampling - trained staff?
Testing
- General- QC system, Flow sheets, Methods, Contract testing, Re-testing
- Testing of raw materials - Testing methods/equipment, testing in the processing areas
- Testing of intermediates - System, sampling
- Testing of final products - System, sampling, samples handling
- Stability testing - System, ongoing, facility, equipment
- Validation of test methods - Policy, Validation process, Validation data, Method transfer
Results and release of test results
- Handling of test results - Transfer of raw data, Laboratory Management System, Summary of raw data, Evaluation of test results, Trending
- Out of Specification (OOS) test results - System/OOS, Laboratory errors, Process/Procedure related errors, Evaluation of Out of Specification results, Test results invalidation, and corrective action
- Failures- Retesting and Resampling - Company's procedure
- Release of test results/ analytical reports/ certification - Process release of test results, Feedback to batch release, Preparation of Analytical report, Preparation and release of Certificate of analysis
Attend the seminar Quality Control Laboratory Compliance - cGMPs and GLPs to examine the fundamental requirements for all QC laboratories subject to FDA inspection, recent trends from FDA inspection reports and enforcement actions.
Ms. Thomas has over two decades of cGMP hands-on industry experience in both pharmaceutical and medical device manufacturing operations. Her experience covers all Quality Systems; as well as, all areas of validation; including, process/product validation, facilities validation, CSV and 21 CFR Part 11, test method validation, equipment/automated processes and cleaning validation. Utilizing strategic thinking, risk-based approaches, and Lean principles, she has demonstrated success in steering and managing complex projects within the pharmaceutical and medical device industries.
FAQs on cGMP and GLP Regulations and Inspections for Quality Control Labs
1. What is the difference between cGMP and GLP in a quality control lab?
- cGMP (Current Good Manufacturing Practice) applies to manufacturing operations, including QC testing for drug products, and is primarily about ensuring consistent quality, purity, identity, and strength.
- GLP (Good Laboratory Practice), on the other hand, refers to nonclinical laboratory studies (e.g. safety/toxicity studies) and is governed by 21 CFR Part 58.
- In QC labs, cGMP ensures that test methods, documentation, and sampling are consistent with manufacturing standards, while GLP ensures data integrity, traceability, and scientific rigor in nonclinical testing.
2. Why do FDA inspectors focus so much on QC laboratories?
- Because quality control labs play a critical role: decisions to release or reject batches are based on their test results.
- During inspections, FDA looks closely at raw data, method validation, instrument calibration, out-of-specification (OOS) investigations, and how labs document every step.
- If a QC lab fails to comply, there’s a real risk that sub-standard or unsafe products could go to market — so regulators treat these labs as high-risk areas.
3. What are the key cGMP requirements for a QC lab?
- The lab should have scientifically sound and appropriate specifications for all materials, test procedures, and sampling plans.
- Instruments must be properly calibrated, methods must be documented and validated (accuracy, sensitivity, specificity, reproducibility), and each test result must be recorded at the time of performance.
- There should be a written sampling plan, and representative samples should be drawn rationally (e.g., random sampling).
- For stability testing, labs must run a formal program (accelerated + long-term), maintain reserve samples, and document everything.
- Lab records must be meticulous: test methods, sample descriptions, validation data, raw data, and conclusions should all be documented.
4. What does GLP (21 CFR Part 58) require from a lab conducting nonclinical studies?
- A Study Director must be appointed for each study — someone educated and experienced who takes overall responsibility.
- There must be an independent Quality Assurance Unit (QAU) that monitors all activities to ensure compliance.
- Labs need written Standard Operating Procedures (SOPs) for all test methods, and these SOPs must be reviewed, revised, and archived.
- Raw data, instruments, and test facilities must be controlled so that the data’s reliability can be verified.
- Any deviations must be documented, and final reports signed by the Study Director, with any changes handled via formal amendments.
5. How can a QC lab ensure data integrity under both cGMP and GLP?
- Implement robust documentation practices: every test, every result, every change must be recorded.
- Use validated test methods: labs should prove that their methods are accurate, reproducible, and fit for purpose.
- Maintain an audit trail: for electronic systems, only authorized people should enter data, changes should be timestamped and justified, and deletion should be prevented.
- Regularly calibrate instruments and verify their performance.
- Conduct periodic internal audits and trend analysis — e.g., on out-of-specification results, deviations, or rejections.
6. What common pitfalls or compliance issues do QC labs face under these regulations?
- Incomplete or poorly documented SOPs or methods.
- Inadequate sampling plans — not using statistically rational sampling or poor sample representativeness.
- Failing to validate test methods properly (e.g., insufficient data for reproducibility or specificity).
- Poor handling of OOS (Out-of-Specification) results — labs may not investigate rigorously or document corrective actions.
- Weak data integrity controls in electronic systems (unauthorized edits, no audit trail).
- Not having a strong Quality Assurance Unit in GLP studies, or not following GLP structure (like Study Director or QAU independence).
7. How frequently do regulators inspect QC labs for cGMP and GLP compliance?
- For cGMP: FDA inspects QC labs as part of broader facility inspections, often focusing on labs that release batches.
- For GLP (21 CFR Part 58) studies: GLP labs are subject to inspections to verify that data from nonclinical studies are reliable, and that all GLP elements (QAU, SOPs, raw data) are in place.
- Inspection frequency can vary, but labs should be ready at all times — robust internal systems, audits, and documentation make compliance sustainable.
8. Can a QC lab be accredited for GLP compliance?
- While the FDA does not “certify” labs for GLP, labs often align with or demonstrate competence via third-party accreditation based on ISO standards.
- Some accreditation bodies (e.g. ANAB) offer programs specifically tailored to ISO/IEC 17025 plus GLP (21 CFR Part 58) compliance.
- Accreditation helps labs show that they meet both regulatory (GLP) and quality management (ISO) standards — boosting credibility for clients and regulators.
9. What are “reserve samples” and why are they important in stability testing?
- Reserve samples are representative portions of a batch set aside for long-term storage under defined conditions.
- They help labs to re-test in future if there are quality concerns, complaints, or unexpected stability issues.
- Documenting reserve samples (how they’re stored, tested, and tracked) is critical for regulatory compliance and product safety.
10. How should a QC lab handle out-of-specification (OOS) test results?
- Investigate thoroughly: determine whether the issue is due to lab error, sample problem, method issue, or manufacturing process.
- Document the investigation, corrective actions, and any re-testing or re-sampling plan.
- Trends should be analyzed — repeated OOS might indicate a systemic problem in methods or processes.
- Ensure that final decisions on batch release incorporate the investigation outcomes, and maintain these records as part of GMP documentation.




