Clinical Trials - Providing relevant information in the IND application
'The federal law requires that a drug be the subject of an approved marketing application before it is transported or distributed across the state lines.' A sponsor who wishes to conduct a clinical trial that involves an investigational new drug (IND), should obtain exemption from FDA to allow the shipping of the investigational drug to clinical investigators in many states. The sponsor files the application to technically obtain the exemption from FDA. This article reviews the IND application types, and the information to be filled in the IND application.

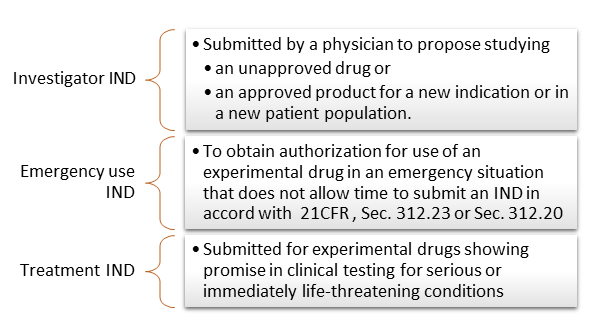
There are 3 IND types:
IND applications are classified into 2 categories
- Commercial
- Research (noncommercial)
It takes 30 working days after the submission of the IND application for the sponsor before initiating any critical trails. FDA reviews the IND for safety to assure that research subjects will not be subjected to unreasonable risk during this time.
3 Broad areas the IND application must contain information about:

Please note that this is a brief overview information required for an IND submission. The webinar 'GMP Expectations for Phase I and First-in-Man Clinical Trials' takes a deeper dive into the subject.
The Information required include for IND application includes:
- Forms for correspondence
- Form FDA 1571 Investigational New Drug Application. Find Instructions for filling out FDA 1571 here.
- Form FDA 1572 Statement of Investigator. Instructions for filling this form are here. Click here for frequently asked questions pertaining to the statement of Investigator.
- Form FDA 3674 Certification of Compliance, under 42 U.S.C. 282(j)(5)(B), with Requirements of ClinicalTrials.gov Data Bank (42 U.S.C. 282(j). Click here for filling Instructions. For guidance click here.
- Note: Companies usually include a cover letter. It adds a human touch.
- Table of Contents (B' 312.23(a)(2))
- Should be detailed for reviewers to find items in the application easily. It is helpful if the location of information is provided by volume and page.
- Introductory Statement and General Investigational Plan (B' 312.23(a)(3))
It is a brief overview of the general investigational plan for the study. This information is repeated later in the IND, in detail.
- Section 1 must include the name of the drug, active ingredients, its pharmacological class, structural formula (if known), and formulation of the dosage form to be used, route of administration, and broad objectives and expected duration of the study.
- Section 2 must include a summary of previous human experience, reference to other INDs, if relevant, and investigational and marketing experience in other countries, if applicable.
- Section 3 must indicate if the drug has been withdrawn from investigation or marketing for any safety or effectiveness reasons, including the location and the reason
- Section 4 must provide a summary of plans for investigating the drug within the next 12 months, including rationale for the study, indications(s) to be studied, general plan for evaluating the drug, kind of studies planned for the first year (specify if these plans are not yet complete), expected number of patients to be enrolled and anticipated risks based on animal toxicology data.
- Investigator's Brochure (B'B' 312.23(a)(5), 312.55)
- Include a copy of the Investigator's Brochure where applicable
- Provide in it a compilation of the clinical and non-clinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects
- It helps the Investigator(s) understand the rationale of key features of the protocol (dose frequency/interval, methods of administration)
- Protocols (B' 312.23(a)(6))
- A protocol is a plan. It tells how a study, either clinical or nonclinical is planned.
- Submit a protocol for each planned study
- Submit a Form FDA 1572 for each Investigator participating in the study
- Protocols not submitted with the original IND must be submitted in an IND Protocol Amendment.
- Chemistry, Manufacturing, and Control Information (B' 312.23(a)(7))
- Information on drug substance, drug product (preparation, manufacturer, components, etc.)
- The data required on chemistry, manufacturing and controls is very similar
- The amount of CMC information that should be provided will vary with the phase of the investigation, the proposed duration of the investigation, the dosage form, and the amount of information otherwise available.
- 'The label for the immediate packaging of the investigational drug, which must contain the statement "Caution: New Drug - Limited by Federal (or United States) law to investigational use" (B' 312.6(a))
- An environmental assessment under 21 CFR 25.40 or a statement requesting a categorical exclusion from an environmental assessment under provisions provided for in 21 CFR 25.31(e) (B' 312.23(a)(7)(iv)(e))'
- Pharmacology and Toxicology Information (B' 312.23(a)(8))
- The first IND submission should include all current pharmacology and toxicology information upon which the decision to proceed to study the product in humans was based, up through what is known when the IND is ready for submission.
- As more information is gathered and the studies progress, submit informational amendments to keep the IND current
- In the toxicology information, include information on the toxicological effects of the drug in animals and in vitro
- Guidance for Industry: Content and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs, Including Well-Characterized, Therapeutic, Biotechnology-derived Products here.
- Previous Human Experience With the Investigational Drug (B' 312.23(a)(9))
- Include relevant information about previous investigations or marketing in the United States and other countries, including published material relevant to the product's safety and/or effectiveness.
- List other countries where the product has been marketed and whether it was withdrawn from any of those markets (and why), or state that there has been no previous human experience.
- Previous human experience may be presented in an integrated summary report.
- Other Important Information (B' 312.23(a)(10)(i) - (iii))
- Other types of important information on special topics as 'Drug dependence and abuse potential' and 'Radioactive drugs' might be required
Guidance documents to help prepare INDs
21CFR Part 312 - Investigational New Drug Application
21CFR Part 314- INDA and NDA Applications for FDA Approval to Market a New Drug (New Drug Approval)
21CFR Part 316- Orphan Drugs
Orphan Drugs- Good Lab Practice for Nonclinical Laboratory [Animal] Studies
21CFR Part 50- Protection of Human Subjects
21CFR Part 56- Institutional Review Boards
21CFR Part 201-Drug Labeling
21CFR Part 54- Financial Disclosure by Clinical Investigators
Documents of Interest to IND sponsors pertaining to Emergency Use of an Investigational Drug or Biologic
Information sheet
Guidance for Institutional Review Boards and Clinical Investigators - How to obtain Emergency IND, Emergency Exemption from Prospective IRB, Approval Exception from Informed Consent, and Requirement Planned Emergency Research, Informed Consent Exception.
Instructions for Sponsors of Emergency Investigational New Drug (EIND) Applications for Antimicrobial Products.
Attend the seminar FDA's 'GMP Expectations for Phase I and First-in-Man Clinical Trials' to understand how to take a comprehensive and systematic evaluation of the manufacturing setting. The seminar will also guide you to take appropriate actions prior to and during manufacturing to eliminate or mitigate potential hazards to safeguard the quality of the phase 1 investigational drug.
The speaker David L. Chesney, MSJ, is the Principal and General Manager for DL Chesney Consulting, LLC, providing GMP and GCP compliance consulting and training services to clients world wide. He has 47 years experience, evenly divided between the FDA and the private sector, including over 20 years as Vice President, Strategic Compliance Services for PAREXEL Consulting. Prior to joining PAREXEL Consulting, he served 23 years with the FDA as an Investigator, Supervisory Investigator, Director of Investigations and ultimately as District Director in San Francisco, managing all FDA operations in Northern California, Nevada and Hawaii.




