Complaint Management and Medical Device Reporting overview for Medical Device Manufacturers
FDA requires Medical device manufacturers to report events that involve medical devices in order to detect and correct problems in a timely manner. Despite the best planning of medical devices, sometimes adverse events occur. These events may include medical device malfunctions, serious injuries, and deaths. Noncompliance may cost may cost millions in fines, product recalls, and litigations.
If you are involved in handling functions involving product complaints, recalls, or medical device reporting, this article will help you minimize complaints and save significant product recall and litigation costs. Complaint management procedures are among the biggest reasons for Form 483 issued to medical device manufacturers. Also, citations related to complaint management procedures are on the rise. Additionally, Complaint handling inconsistencies are often identified by FDA during inspections. Many warning letters issued to medical device manufacturers point in the same direction.
You can mitigate or minimize medical device complaints by managing complaints effectively:

Managing complaints
- Capture complaints you receive from all sources including phone, email, snail mail, forms on the company's website or social media. Make sure that nothing slips through the cracks across multiple channels.
- Create a product complaint report
- Make an adequate investigation of the root cause of the problem
- Clearly document activities and decisions
- Report adverse events to FDA
- Follow up with the complainant
- Communicate with management and departments across the organization
Causes of complaint management failures
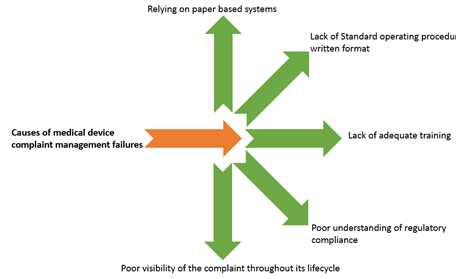
Relying on paper-based systems
Most companies have adapted effective computer systems for complaint management. However, there are small pockets of the industry to adapt computer systems. The vulnerability of using paper-based systems for complaint management is high. Paper-based systems are prone to human errors such as information entry, filing, safety, and other reasons commonly known.
Lack of Standard operating procedures (SOPs) in written format
SOPS are critical to a successful complaint management system. They remove ambiguity and subjectivity. They provide a common definition of various complaint management terminologies, define process owners and expectations, identify processes for complaint handling and management, and define complaint escalation process, timelines in line with regulatory requirements and the company's quality management system. They simplify performance management, and enable continuity of complaint handling.

Lack of adequate training
Personnel who handle complaints must be equipped with thorough knowledge of the regulatory requirements, must know when to report and to whom to report, the timelines of reporting etc. Each product category complaint management requirements vary from the other. Some medical device companies train their personnel on an ongoing basis to ensure that their staff is up-to-date with the latest developments.
Poor understanding of regulatory requirements
Regulatory requirements are ever evolving. Understanding the requirements and keeping abreast with the evolving changes is very challenging. The complexity of reporting required by regulatory agencies in global markets is a major challenge.
Poor visibility of the complaint throughout the lifecycle
Many departments are usually involved in managing complaints. Hand-offs and back and forth of complaint management from one person to another within the organization, managing the date of awareness can be challenging unless there is a robust system in place. The regulatory clock starts ticking from the day the Company becomes aware of the complaint (More details on this in fig 2 given in the later part of this article.).
As stated at the outset of this article, despite a robust complaint management system, problems may occur. FDA requires Medical device manufacturers to report events that involve medical devices in order to detect and correct problems in a timely manner. Building a winning medical device reporting team and the process is paramount to the success of your complaint management program.
The following part provides an overview of the FDA's medical device reporting guidance for manufacturers.
A brief overview of Medical Device Report (MDR) guidance
The MDR requirements are found at Title 21 of the code of federal regulations part 803. It implements section 519 of the Federal Food, Drug, and Cosmetics Act. It sets forth the report and record keeping requirements for certain medical device related adverse events for device user's facilities, manufacturers, and importers.
Guidance for meeting the requirements is found in the Medical device reporting for manufacturers document issued on November 8, 2016.
The section 2 of the MDR guidance describes in great detail what types of events must be reported to FDA.
- Events where a manufacturer suspects that one of their medical devices may have caused or contributed to a death or serious injury.
- Certain malfunctions even if the event does not involve a patient.
The section 3 describes additional requirements apart from reporting adverse event. These include:
- Conducting a complete investigation of each event or complaint they receive
- Reporting all information in their possession relevant to the adverse event
- Having written procedures for adverse event management
- Implementing the written procedures
- Establishing and maintaining MDR event files
- Ensuring that there is a system in place to provide access to that information for FDA inspection to inspect the facility
A medical device may not be the cause, it may have contributed to the death or serious injury. How can the manufacturers make discretionary judgments to determine if the death or serious injury involved their medical device? Examining Results from Top Medical Device Observations During an Inspection will be a great source to making discretionary judgments.
The section 4 covers specific issues and situations.
The section 5 provides guidance about the logistics of how to file a report, and specifications for electronic submission of report to FDA.
Who reports adverse events, to whom, and within what timeframes?

To minimize risk of regulatory enforcement actions pertaining to Post-Market activities, Complaint Handling, MDRs, and Recalls, attend the seminar 'Managing Your Complaints and Obstacles in Post-Market Requirements -- Results from Top Medical Device Observations During an Inspection'. This Seminar will help you stop spinning your wheels with nonessential activities, and leave you with a comprehensive learning package that only Rita Hoffman, a former FDA CDRH Recall Branch Chief with experience across the device, drug and veterinary industries can provide.




