COTS software validation, regulatory requirements, and risk analysis methodologies
The regulated industry has developed requirements and standards for managing systems and processes related to product design, development, manufacturing, packaging, distribution, and monitoring. Software vendors design and develop systems to ensure they meet the industry standards. The commercial-off-the-shelf (COTS) software developed and supplied by software vendors must undergo validations by end users. A consistent process to validate these systems is invaluable to companies who use COTs software

Why explore COTS validation
Software vendors who provide Software as a Service (SaaS) and cloud computing IT resources are not regulated. Therefore regulated companies that outsource must ensure compliance for both infrastructure qualification and computer system validation to avoid FDA form 483s and Warning Letters. Understanding the computer system can help regulated companies, software vendors and Saas/cloud providers. This article sheds light.
- The CSV course Computer System Validation - Reduce Costs and Avoid 483s will explore proven techniques for reducing costs associated with implementing, using, and maintaining computer systems in regulated environments.
Computer system validation is a regulatory requirement
- FDA Guidance on Part 11 Scope and Application states - We recommend that you base your approach on a justified and documented risk assessment and a determination of the potential of the system to affect product quality and safety, and record integrity.
- clause 2 [5] of EU GMP Annex 11 states The extent of validation necessary will depend on a number of factors including the use to which the system is to be put, whether the validation is to be prospective or retrospective and whether or not novel elements are incorporated.
EU GMP Annex 11
- 5.40: GMP related computerised systems should be validated. The depth and scope of validation depends on the diversity, complexity and criticality of the computerised application.
- 5.41: Appropriate installation qualification and operational qualification should demonstrate the suitability of the computer hardware and software to perform assigned tasks.
- 5.42: Commercially available software that has been qualified does not require the same level of testing. If an existing system was not validated at the time of installation, a retrospective validation could be conducted if appropriate documentation is available.
FDA Quality System Regulation (21 CFR Part 820
Applies to medical device industry:
- Design Controls: Section 820.30(g) Design validation. Each manufacturer shall establish and maintain procedures for validating the device design. Design validation shall ensure that devices conform to defined user needs and intended uses and shall include testing of production units under actual or simulated use conditions. Design validation shall include software validation and risk analysis, where appropriate.
- Production and Process Controls: Section 820.70(i)
Automated processes. When computers or automated data processing systems are used as part of production or the quality system, the manufacturer shall validate computer software for its intended use according to an established protocol. All software changes shall be validated before approval and issuance. These validation activities and results shall be documented.
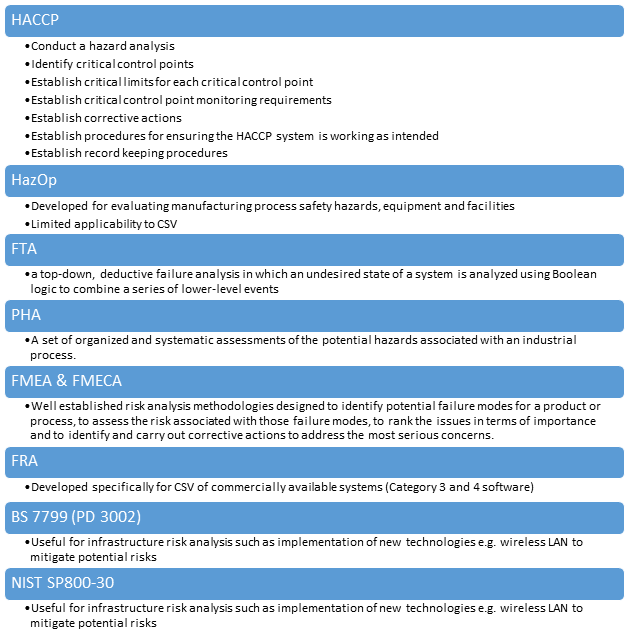
Risk analysis methodologies
Risk assessment methodology
There are many risk assessment methodologies for computer system validation of COTS software. The following is one of the risk assessment methodologies.
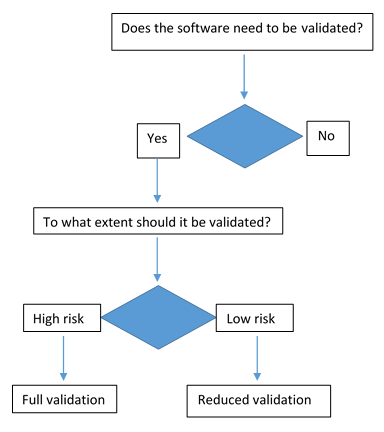
Factors determining the risk factors
Validation coverage should be based on the software's complexity and safety risk not on firm size or resource constraints.
Risk assessment based on system usage:
GAMP Part 11 Good Practice Guide lists some examples.
- High risk:
- Data that are submitted directly to regulatory agencies or are included in regulatory submissions
- Data supporting batch release (e.g. Certificate of Analysis) of drug product, clinical trial material or Active Pharmaceutical Ingredient (API)
- Stability data for drug products
- Data from or support to non-clinical laboratory studies
- Clinical trial study data
- Laboratory support to clinical studies
- Low risk:
- In-process monitoring of drug product and APIs
- Supportive data not directly submitted to regulatory agencies
- Pharmacology data
- In vitro data
- Research data
- Data generated in the development of analytical methods
Risk assessment based on the nature of the system
The higher the GAMP category, the higher the risk to records. The more unique the software is the less it is tested is the rationale.
High risk
COTs software package that involves configuring predefined software modules and possible developing customized modules (GAMP category 4).
Low risk
COTs standard non-configurable software package providing an off-the-shelf solution to a business or manufacturing process (GAMP category 3)
If you are a regulatory, clinical, and IT professionals working in the health care, clinical trial, biopharmaceutical, and medical device sectors, attend the seminar Computer System Validation - Reduce Costs and Avoid 483s, to understand the latest computer system industry standards for data security, data transfer, audit trails, electronic records and signatures, software validation, and computer system validation.
The speaker, David Nettleton is a Computer System Validation's principal, an industry leader, author, and teacher for 21 CFR Part 11, Annex 11, HIPAA, software validation, and computer system validation.




