How to Ensure Safety and Efficacy of your Medical Device throughout its life-cycle with Risk Management
Medical Devices that are manufactured for human applications like treatment and diagnosis must be safe and effective. Such devices include instruments, an apparatus, or a material that are used for daily patient care or medical scientific purposes. Manufacturers responsible for developing new devices should take adequate precautionary measures to ensure that the devices don't cause hazards when people use them. They are responsible for:

- Identifying hazards that are related to the device
- Estimating and assessing the related risks
- Controlling the identified risks
- Evaluating if the residual risks are acceptable
- Regularly monitoring and reviewing new data with regard to these risks
This article discusses the key role Risk Analysis, Risk Management Process, Application of Risk Management tools plays in medical device design development and the advantages of Risk management analysis. The aim is to help develop medical devices that are safe and effective for use throughout their product life-cycle and to help evaluate new device submission and design control documentation.
Tip: While Medical Devices, Pharmaceuticals, and Combination Products have had unique histories for the evolution of risk management, the management of compliance risk is a common business process for all three areas.
The FDA has provided guidance on particular risk analysis approaches and procedures like fault tree analysis (FTA) or failure mode and effects analysis(FMEA). By starting to think about risk at the earliest possible in the device or process development, and reviewing those problems in an organized manner throughout the development process, medical device manufacturers can manage and reduce risk effectively.
For a review of Quality Risk Management for Pharma, Biopharma and Combination Products - ICH Q9/ISO 14971 attend this seminar.
- Risk Management Overview
- Risk analysis is a regulatory requirement
- Ensures that the medical device is safe
- It helps save unnecessary costs associated with recalls through early identification of design problems.
- Risk analysis must be included in regulatory submissions
- It is ethical
- Provides a measure of protection from product liability damage awards
- To make sure that the medical device is safe and effective for use
- To prevent unsafe devices from entering the market
- It enhances the credibility of the manufacturer
- Document definitions of what is to be done and the way to do it
- Define duties and accountability. Also, define what requires authorization.
- Define the qualifications, skills, and knowledge required to implement the process and what is made available to train those without those skills.
- Create and maintain documents to show adherence to policies and procedures
- Implement steps to validate and confirm that the procedures are adhered to
- Ensure that procedures are in place and operate correctly.
- Inherent safety by design
- Protective steps in the device or its manufacture
- Safety instructions like warnings, maintenance schedules, and more.
- Inherently safe design
- Preventive controls like default operating modes
- Soft controls like labeling, training, and operator instructions
- Corrective actions in a timely manner - the right response for the events like a patient-specific response
- Prevent or remove failure causes
- Identify or discover the failure at the earliest
- Minimize the impact of failure
- Toxicity
- Flammability
- Reactivity of raw materials and wastes
- Sensitivity to environmental factors such as temperature and humidity
- Mechanical or electronic hazards
- Human factors associated with the operator-device interface
- Patient-device interface for effective delivery of energy, administration of drugs or control of life-sustaining functions
- Procedure Analysis
- Malfunction of the software component
- In the event that the software component fails to follow its schedule (time) to function
- In the event, the software component operates using wrong or corrupt data
- ISO 14971:2007
- Harm: physical injury or damage to health of person, or damage to property or environment
- Hazard: potential source of harm
- Hazardous situation: circumstance in which people, property, or the environment are exposed to one or more hazard(s)
- Severity: measure of the possible consequences of a hazard
Why is Risk management essential?
The first step to effective risk management is hazard identification. By assessing the potential results of the risks and the probability of their occurrence, the degree of risk can be projected. This value is matched to the company's risk-acceptability levels. If the levels are unacceptable, the risks should be corrected before proceeding any further. The necessary steps are in figure 1 as shown below:
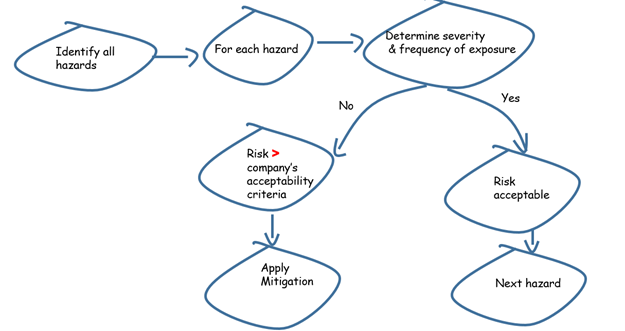
Figure 1.Risk management process
Risk can be minimized to acceptable levels but cannot be removed completely. Hence, the risk that remains after taking adequate steps must be managed.
Risk control
The process of arriving at conclusions and the implementation of preventive steps for minimizing and maintaining risk within acceptable levels.
Risk control and monitoring tasks
The activities purposed to remove or minimize every risk to satisfy the predetermined risk acceptability levels. The implementation may include one or more risk control activities. Risk controls may start at the initial design input stage and carry on over the product life-cycle. Some regulatory guidelines recommend a predetermined order of risk controls viz.
All through the device's life-cycle, the risks should be monitored to ensure that it is maintained within acceptable levels. Also, monitoring throughout the life-cycle is required to identify if there are new hazards or risks.
A well-defined quality management system consisting of elements like production, complaints, customer feedback is an invaluable resource for effective monitoring.
Risk control measures
Residual risk (RR)
Risk remaining after risk control measures have been taken
Is the overall RR of the device acceptable?
Does data suggest that the medical benefits of the intended use outweigh the overall residual risk?
If the evidence supports decisions that medical benefits prevail over overall RR, then overall RR can be judged acceptable. Else, the overall RR remains unacceptable
Risk mitigation
Risk mitigation is the measures you are going to take about the situation to:
Risk mitigation Identifies what safety risk control measures have been taken to minimize the probability and severity of the resulting harm. These could include preventive measures taken in the design, processes, and or/labeling.
Hazard analysis
Prior to developing the final design, an initial hazard analysis can be performed to form a hazards starting-point related to a device. Basically, the analysis includes a record of main components and operating requirements of the device and assessing their potential risks. The listing of the main components and operating requirements could include raw materials and wastes, hardware, monitoring and control systems, human-device interfaces, services, and the operating environment.
What may need to be assessed for potential hazards?
Deep thinking and brainstorming are required before performing the initial hazard analysis. Think hard to dive deep with an open mind, and brainstorm to list every possibility of the occurrence of a hazard and the severity of the hazard. Such an approach can help you identify failures, assess potential consequences, and develop risk management strategies while lowering the design cost.
Although the hazard likelihood details are insufficient for assessment at this stage, you can use similar devices and their histories to make comparisons. With deep thinking and comparisons, you may identify the need to make drastic changes in the conceptual design. As the conceptualization phase is the initial phase, it allows substantial flexibility. The aim is to remove all high-severity hazards and minimize low-severity hazards to the best possible.
While developing the prototype, a more comprehensive hazard and risk analysis can be conducted. This stage of design allows availability of process and mechanical drawings and the defined basic processes. There are many types of analysis techniques to review the device and its operation like top-down and bottom-up approaches.
It is a good practice to conduct a separate analysis focused on procedures. Every step in the process is assessed to decide if a wrong procedure could adversely affect the outcome.
Checklists are great tools for performing design reviews. However, they are usually not enough. The checklist must be comprehensive to cover all potential hazards failing which the design reviews would be incomplete.
Risk analysis must consist of any risks related to the manufacture and delivery of the device to its purposed location. If the devices consist of solutions and components that degrade due to environmental factors such as cold, light, humidity, heat etc., the storage and transportation procedures need should be reviewed. Such a review may guide you to change the packaging or warning on storage or packaging containers.
If changes are made during the design process, they should be reviewed to make sure that the hazards don't find a place in the design. Just before starting the production, a final review should be performed. Apart from using extensive checklists, a physical review of the device and its workstation should be conducted to identify any issues that can't be identified from written records or drawings. The findings and observations of the design review and the final action items that must be completed before beginning the production must be documented and prioritized.
Crucial to review is the software that is used to control or monitor a medical device. By grouping the software into its important functions, you can ease the review process.
Software malfunctions can bring in undesired results, specifically those that use corrupt data. Crucial to minimizing software errors is to have the means to find the effects of software errors on a medical device.
A standard for risk management companies use to show that they are compliant with the Risk management requirements for Medical Devices Directive. (MDD). It provides a framework for risk management process. The key requirements are discussed here.
Definitions
Attend the seminar Compliance Risk Management in Medical Devices, Pharmaceuticals, and Combination Products to get answers to key questions regarding Risk Management and Product Risk Management, the acceptability criteria for Compliance Risk Management, the expectations for managing risk in Pharmaceuticals (cGMP), and the interface of evolving landscape of Compliance Risk Management with with the evolving landscape of Combination Products.
The speaker Stan Mastrangelo has over 30 years of professional work experience in Quality Assurance of medical devices, pharmaceuticals, and foods. Stan has held positions such as Senior Quality Engineer, Corporate Quality Assurance Auditor, Plant QA Manager, QA Director, and Consultant. Stan was a member of the ANSI Executive Standards Board. Stan has had extensive involvement in the development of International Risk Management Standards. Stan was a member of the ISO Joint Working Group for Risk Management of Medical Devices (that developed ISO/IEC14971). Stan was a committee liaison to the ISO Technical Management Board Joint Working Group on Risk Management that developed ISO 31000 which is the Risk Management Standard for all sectors. Stan was on the US PhRMA (Pharmaceutical Research and Manufacturers Association) Team that supported the development of ICH (International Conference for Harmonization) Standard Q9 titled Quality Risk Management for Pharmaceuticals. Stan also served on various IEC Standards Teams related to IEC 60601, IEC 80001 and Risk Management in the Software Lifecycle. Stan is an Adjunct Professor at Virginia Tech and was a co-developer of a Masters Degree Program in Medical Product Risk Management. Stan is on the Risk Management Committee for the IECEE.




