Importing Medical Devices - 5 Key Factors to Ensure Regulatory Compliance
- Establishment is registered with the FDA
- Devices have been listed with the agency
- Devices are manufactured in compliance with FDA's Quality System Regulations or QSR
- Adverse events related to medical devices are reported in a compliant manner
- Premarket Notification (510(k)) or Premarket Approval have been obtained from the FDA for any device to be imported into the US
- Foreign manufacturers should have a designated agent in the US.
- Assesses and collects all duties, taxes, and fees on imported merchandise
- Administers and reviews import entry forms
- Enforces CBP and related laws
- Repackage the product
- Modify the container, wrapper, or labeling of the device or device package
- Medical Device Reporting (MDR) under 21 CFR 803
- Reports of Corrections and Removals under 21 CFR 806
- Medical Device Tracking under 21 CFR 821 if applicable
- Designate a US Agent
- Only one US agent per establishment is allowed
- This agent can be designated as the foreign manufacturer's official correspondent
- At the time of registration, the foreign manufacturer should provide the FDA the name, address, telephone and fax numbers, and e-mail address of the U.S. agent
- Either reside in the US or maintain a place of business in the US
- Have a proper address - a PO Box number won't be enough
- Avoid using an answering service - the FDA expects the telephone number provided to be answered by the agent or its employee during normal business hours
- Assisting the FDA in communications with the foreign manufacturer
- Responding to the FDA's queries regarding devices to be imported or already imported into the US
- Assisting the FDA in scheduling inspections of the foreign manufacturer
- Receive any documents, letters or other communication that the FDA wants to send to the foreign manufacturer
- Ensure Establishment is Registered with the FDA
- At the time of registration, the foreign manufacturer must provide a list of devices that they intend to import into the US and the activities performed on those devices.
- The FDA premarket submission number (510(k), PMA, PDP, HDE) should be provided.
- Foreign manufacturers must identify all known US importers of their devices
- Initial importers must identify the manufacturers of the devices they are importing.
- All proprietary names under which a device is marketed must be reported, at a minimum, when a device is first listed and during the annual update of registration and listing information.
- Provide All Necessary Product Information at Time of Entry
- Respond with Evidentiary Support or Plan to FDA Notice of Action
- Recondition the Device to Bring It into Compliance
Comply with FDA regulatory requirements to ensure a trouble-free medical device import process.
The FDA strictly regulates the import of medical devices into the United States. It has detailed regulatory requirements for foreign medical device manufacturers that should be followed if imported devices are to be sold in the US market. Non-compliance with these rules can result in detention of products at ports of entry, unnecessary expenses involved in paperwork, queries and hearing and a loss of revenue for the importer. This article provides five best practices that both foreign medical device manufacturers and importers in the US can follow in order to ensure that the device import process is quick and efficient.

Regulations Governing Import of Medical Devices
The FDA does not recognize regulatory approvals obtained in other countries, therefore foreign firms that manufacture medical devices imported into the United States need to comply with applicable US regulations before doing so.
Importers should ensure that they comply with the following:
In addition to FDA regulations, the importers must also comply with Customs and Border Patrol (CBP) requirements. The CBP's role in the import process is to administer the Tariff Act of 1930. The CBP also does the following with respect to medical devices being imported into the US:
Who is An Initial Importer?
According to the FDA guidelines, an initial importer is any importer who markets an imported medical device to the person who makes the final delivery or sale of the device to the ultimate consumer or user. The initial importer does not:
Initial importers have to comply with the following FDA regulatory requirements:
The Medical Device Import Process
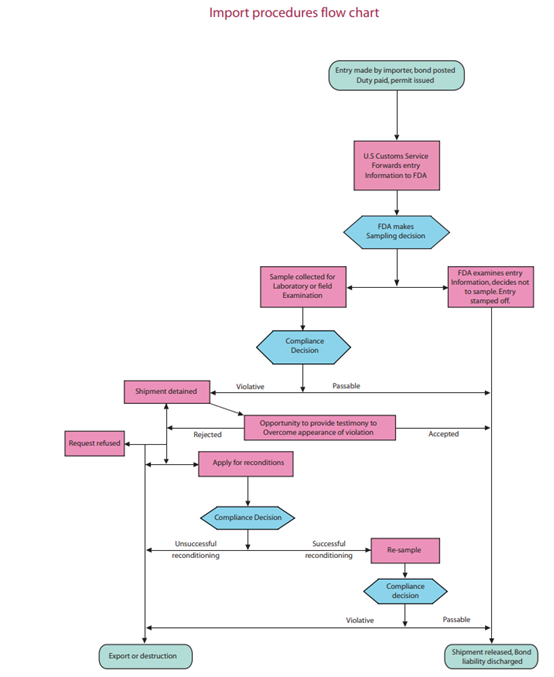
The import process begins when the importer or filer submits the necessary entry information to the local CBP district office. The FDA's import process is illustrated in the import procedures flowchart as shown below:
Consequences of regulatory non-compliance
If FDA-regulated items do not conform with FDA regulations and recommendations, they may be denied entry into the United States. Over 1,000 FDA-regulated goods were denied entrance into the United States in August 2020 alone.
The detention of a product, as well as the subsequent actions involved in obtaining its release, necessitate a significant financial investment. If the importer fails to successfully appeal the FDA's detention during the informal hearing, the merchandise must be exported or destroyed within 90 days. Failure to do so within 90 days may result in the issue of a Customs Redelivery Notice and a liquidated damages charge of up to three times the lot's value.
5 Best Practices for Ensuring Medical Device Import Compliance
As mentioned earlier in this article, foreign medical device manufacturers importing products into the US have to designate a US agent. The details of a company's US agent should be provided to the FDA at the time of establishment registration. This information is submitted electronically through the FDA Unified Registration and Listing System (FURLS).
Foreign manufacturers should note that:
At the time of hiring a US agent, foreign manufacturer should remember that the FDA requires US agents of foreign companies to:
A US agent's responsibilities include:
Foreign manufacturers should be aware that the US agent is not responsible for adverse event reporting or submitting 510(k) premarket notifications. That is the responsibility of the foreign manufacturer alone
Medical device manufacturers - whether they be foreign or domestic - marketing and selling products in the US have to register their establishment with the FDA on an annual basis. At the time of the annual registration, a user fee also has to be paid to the FDA.
The following conditions must be complied with:
Information provided at the time of entry should identify the product and include appropriate information to demonstrate that the product is in compliance with FDA regulations. Product information should include device name and product code. The product code provided to CBP must include a two-digit prefix identifying the medical specialty in addition to the three letter code.
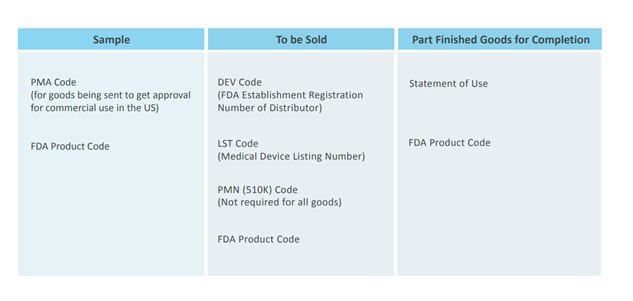
The commercial invoice accompanying the imported medical device will need the following details, depending on whether the product is a sample, to be marketed and sold or is a part finished good for completion:
If the FDA determines that an imported device is out of compliance with regulatory requirements, the agency will issue a "Notice of FDA Action" specifying the nature of the violation to the owner or consignee.
The owner or consignee can ask to have an informal hearing in which testimony regarding the admissibility of the product can be provided. If the evidence submitted does not satisfy the FDA or the owner/consignee is unable to provide a plan that shows it can bring the device into compliance, the FDA will issue another Notice of Action refusing admission to the product.
It is therefore vital that owners/consignees importing the device pay attention to the violations mentioned in the first Notice of Action and give complete evidence showing device compliance or a detailed plan showing how the device will be made compliant.
The initial FDA Notice of Action highlighting violations in medical devices doesn't mean that the importer does not have further avenues for appeal. The FDA will under certain conditions allow the imported to re-label or recondition the contentious device to bring it into compliance with regulations. The importer can request for authorization to carry out such a procedure and if the agency permits it, go ahead with the same.
The FDA will re-examine the device once re-labeling or re-conditioning is completed. In case the device is now found to be compliant, the lot will be released to the importer and a notice usually identified as "Once Detained and Now Released" is issued.
Importers must keep in mind however that reconditioning is allowed only once - so it is important to get it right the first time.
Pay Attention to the Fine Print When Importing Medical Devices
Give the FDA's reputation as one of the most stringent regulatory agencies in the world, foreign manufacturers and US agents/initial importers should ensure that the device they are bringing into the country is compliant with all the necessary regulations. The costs of non-compliance can be huge - in terms of monetary losses and reputational damage as well. Therefore, it is best to read the fine print included in regulatory requirements and avoid unnecessary delays and expenses that detention of devices, appeals and hearings can entail.
How can medical device compliance training help?
The areas highlighted above are just a small part of the wide range of practices and processes for medical device compliance. Subjects such as device compliance are multi-faceted and complex and can be better understood after attending a training course such as the ones offered by ComplianceOnline. Our courses are available as live webinars, training recordings and seminars. We also offer customized training courses developed in conjunction with organizations that wish to train large groups of their employees.
We offer training in other areas device compliance such as the QSR, 510(k), PMA, and other FDA regulatory requirements.
If you need customized training courses or specialist medical device compliance consulting services, please contact us through email [email protected] or call us at this toll-free number: +1-888-717-2436.




