Medical Device Regulatory Compliance - FDA, EU MDR
If you are looking at bringing your new medical device to the market, many factors determine its length of time. However, most important is the time you invest in meeting the regulatory compliance requirements.

- Non-invasive medical devices
- Invasive medical devices
- Active medical devices
- Special category - includes contraceptive, disinfectant, and radiological diagnostic medical devices
- Class I - Non-sterile or no measuring function (low risk)
- Class I - Sterile and a measuring function (low/medium risk)
- Class IIa (medium risk)
- Class IIb (medium/high risk)
- Class III (high risk)
- Preliminary safety testing
- Risk estimation
- Hazard identification
- Hazard management planning
- Risk mitigation strategy planning
- Risk control analysis
- Risk control effectiveness verification
- Overall residual risk analysis
- Risk/benefit analysis
- Final risk management review
- Establishment Registration & Medical Device Listing - 21 CFR Part 807
- Premarket Notification 510(k) - 21 CFR Part 807 Subpart E
- Premarket Approval (PMA) - 21 CFR Part 814
- Investigational Device Exemption (IDE) for clinical studies - 21CFR Part 812
- Quality System (QS) regulation 21 CFR Part 820
- Labelling requirements - 21 CFR Part 801
- Medical Device Reporting (MDR) - 21 CFR Part 803
- General medical devices: The Regulation on Medical Devices 2017/745
- In vitro diagnostic medical devices (IVDs): The Regulation on In Vitro Diagnostic Medical Devices 2017/746
FDA or EU MDR?
As a medical device manufacturer, you are confronted with an important decision - whether to seek the approval of the US Food and Drug Administration or comply with the EU Medical Device Regulation (MDR). This article provides the key differences between the two and provides the step-by-step process for approval.
| FDA | EU |
|---|---|
| Definitions | |
| The FDA approves medical devices for market use based on Title 21-CFR Quality System Regulations. Companies must assess the overall risk profile for each device, follow the prerequisite regulatory criteria and ensure reasonable safety and efficacy while conforming with the corresponding market pathways. | The EU MDR is much more inclusive. It specifies what the responsibilities of the economic operators are, what the revised CE marking process is, the device registration requirements, and their economic operators. It even details the key obligations and conditions for notified bodies, presents post-market surveillance of authorized medical devices, specifies confidentiality and funding responsibilities, provides details about data protection topics, establishes penalties, and sets out the rules for cooperation between member states. |
| Classification of Medical Devices | |
| As the first step to FDA submission, you must identify the class of your medical device by using the FDA's medical device database.
Class I and Class II Device: Devices with low and moderate risk are classified as Class I and Class II. Regulated by the 510(k) notification, product developers of devices that fall under these classifications must be present with data that the device works similarly as a device approved previously. Class III Device:'Class III devices are those that support or sustain human life, are of substantial importance in preventing impairment of human health, or which present a potential, unreasonable risk of illness or injury. Due to the level of risk associated with Class III devices, FDA has determined that general and special controls alone are insufficient to assure the safety and effectiveness of Class III devices. Therefore, these devices require a premarket approval (PMA) application under section 515 of the FD&C Act in order to obtain marketing approval. .. PMA is the most stringent type of device marketing application required by FDA. Some Class III preamendment devices may require a Class III 510(k). (Learn more about Medical Device Premarket Approval (PMA) here) |
The EU MDR has four device categories.
These classifications are based on the risk which determines the level of data and depth of evaluation required. Following are the classifications. Requirements for testing are different for every device class. Also, auditing and validation will be done for devices with preinstalled software: |
| Clinical Evaluations | |
| Premier Research defines clinical in the context of medical device market approval as 'the methodologically sound ongoing procedure to collect, appraise, and analyze clinical data pertaining to a medical device. Clinical evaluation also includes analyzing whether or not there is sufficient clinical evidence to confirm compliance with relevant essential requirements for safety and performance when using the device according to the manufacturer's instructions for use.' | |
| As the submission of most devices to the FDA fall under the 510(k) application, the duration of clinical testing takes about 6 to 15 months based on the purpose of the device and the device type. After assigning the device classification, the following evaluations are conducted for 510(k) submissions.
|
Class I Devices: Evaluations are done based on Annex IV and V and are exempted from conformity assessment to CE marking. Class I devices with medium risk and Class IIa devices may need to undergo conformity assessments based on Annex XI of the MDR (Part A).
Class IIb and Class III devices require robust technical documentation and comprehensive risk evaluation during conformity assessments with notified bodies, and if they are implantable devices the MDR designates them for additional testing under special provisions. |
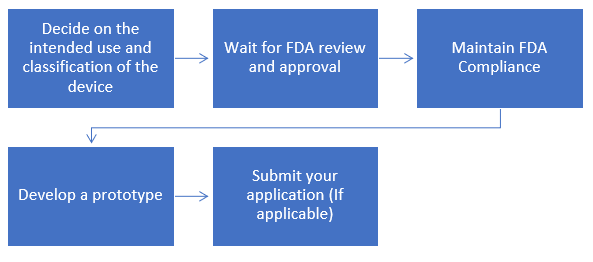
Step-by-step Process to get FDA Approval for your Medical Device
Where ever justified, the 510(k) submission process is the preferred method for obtaining marketing clearance in the U.S. How are such decisions arrived at? Differences from the EU MDD Annex IX schema. What is involved? Sequence of events? FDA expectations. What's included? Desired format? Expected time frames and the post-submission review process "give-and-take". This webinar provides the U. S. FDA Medical Device Approval Process.
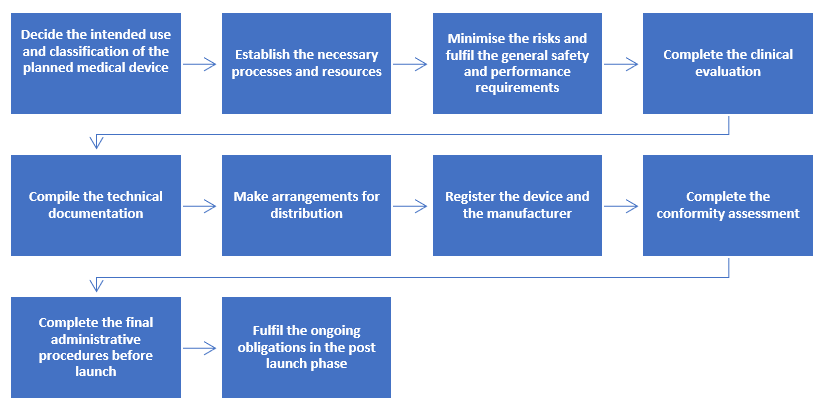
Step-by-step Process to Comply with Regulation (EU) 2017/745
FDA Regulations for Medical Devices
EU Regulations
Looking for related training? Check out our Seminars and Webinars for FDA-regulated Companies.




