Addressing the Medical Device Software Challenges by understanding FDA's Software Regulation Strategy
Many Medical Device Software users face a host of challenges. These include Software failure, malicious remote hacking, and interoperability issues. As the software gets more sophisticated, fixing the software malfunction becomes more difficult. When such malfunctions do occur, it is difficult to decide who is responsible for managing and fixing software problems. How can you prevent or overcome these commercial obstacles and be compliant?

Remaining current with medical device technological tools and strategy can help. Firms should be competent in the application of tools and strategies to ensure software functionality, risk identification, software protection, problem detection, and response strategy. This article provides the required knowledge to address medical device software issues including
- How to classify your software within the three classes the risk-based device classification system as established by the Federal law
- What Risk controls should be applied to the software
- FDA's guidance for medical device software
- How to build a cybersecurity culture
- How to identify interoperability issues
- The scope of FDA's mobile apps regulation
- How to get deeper insights into the topic
Understand and apply FDA classification/risk controls for software in medical devices
Classification
The first step is to place your software into one of the three classes the risk-based device classification system for medical devices as established by the Federal law (Federal Food, Drug, and Cosmetic Act, section 513). The regulatory controls for each device class include:
- Class I (low to moderate risk): general controls - Class I are defined as non-life sustaining. These products are least complicated and their failure possess little risk
- Class II (moderate to high risk): general controls and Special Controls - Class II are more complicated than class I, though they are also non-life sustaining. These are subject to any performance standards.
- Class III (high risk): general controls and Premarket Approval (PMA) - Class III are those that sustain or support life. Their failure is life-threatening.
Think carefully about your classification and document the logic behind your decision.
Risk Controls
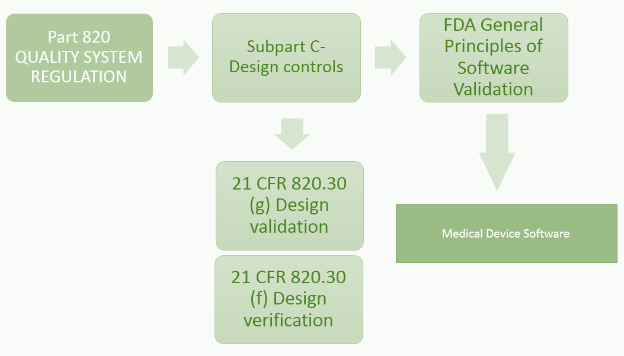
21 CFR Part 820.30 (c) Design Control
Design control covers seven key areas and may be applied to any product development process:
- Users' needs
- Design input
- Design process
- Design output
- Reviews
- Verifications
- Validations
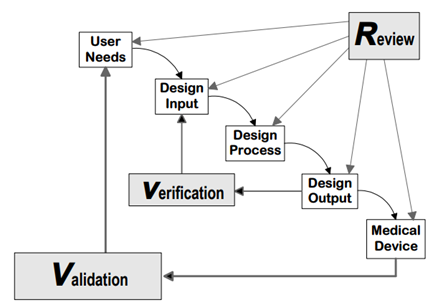
Source: FDA Design control guidance for medical device manufacturers
21 CFR 820.30 (g) Design Validation
'Each manufacturer shall establish and maintain procedures for validating the device design. Design validation shall be performed under defined operating conditions on initial production units, lots, or batches, or their equivalents. Design validation shall ensure that devices conform to defined user needs and intended uses and shall include testing of production units under actual or simulated use conditions. Design validation shall include software validation and risk analysis, where appropriate. The results of the design validation, including identification of the design, method(s), the date, and the individual(s) performing the validation, shall be documented in the DHF.'
820.3(z) Validation means confirmation by examination and provision of objective evidence that the particular requirements for specific intended use can be consistently fulfilled.
- Process Validation means establishing by objective evidence that a process consistently produces a result or product meeting its predetermined specifications.
- Design Validation means establishing by objective evidence that device specifications conform with user needs and intended use(s).
21 CFR 820.30 (f) Design Verification
'Each manufacturer shall establish and maintain procedures for verifying the device design. Design verification shall confirm that the design output meets the design input requirements. The results of the design verification, including identification of the design, method(s), the date, and the individual(s) performing the verification, shall be documented in the DHF'
820.3(a) Verification means confirmation by examination and provision of objective evidence that specified requirements have been fulfilled.
There are 3 levels of software design verification
- Software unit testing: Testing performed to verify the implementation of the design for a single software element or a group of software elements
- Software integration testing: A systematic progression of testing in which many subsections of the software and/or hardware are integrated and tested separately from the rest of the system. This testing continues till the complete system is integrated.
- Software system testing: A testing performed to verify if the fully integrated system meets its specified requirements.
A tougher U.S. FDA expects a firm to maintain certain documents in equipment, process and product software V&V. Drafting a Software Verification and Validation Report Package and Protocol requires a thorough understanding of the process.
A tougher U.S. FDA expects a firm to maintain certain documents in equipment, process and product software V&V. Drafting a Software Verification and Validation Report Package and Protocol requires a thorough understanding of the process.

Understand FDA's Guidance for software
- Off-the-shelf (OTS) software
- Cybersecurity for networked devices containing OTS
- Postmarket cybersecurity management
- Software validation
- 510(k) and PMA submissions for software
- Decision support software - Clinical and Patient Decision Support Software
Build a Cybersecurity culture
Medical Devices security increasingly depend on computers, software, and networking and are often incorporate third party software.
'As part of the software validation and risk analysis required by 21 CFR 820.30(g), software device manufacturers may need to establish a cybersecurity vulnerability and management approach, where appropriate.'
Cybersecurity risk program
Pre Market
'FDA recommends that manufacturers consider the following:
- Identification of assets, threats, and vulnerabilities
- Assessment of the impact of threats and vulnerabilities on device functionality and end users/patients;
- Assessment of the likelihood of a threat and of a vulnerability being exploited
- Determination of risk levels and suitable mitigation strategies; and
- Assessment of residual risk and risk acceptance criteria.'
Device design features that mitigate cybersecurity risk. The subset software documentation should include:
- Software description
- Hazards
- Requirements
- Design specifications
- Traceability
- Development environment
- Verification and validation
- Unresolved anomalies (vulnerabilities)
Post Market
- Engage in post-market surveillance and information sharing and analysis organizations
- Assess the device impact and clinical impact of vulnerabilities and exploits
- Address the risk, actions should commensurate with the risk
- Disseminate, incorporate and iterate
Produce objective evidence that includes:
- Policies
- Procedures
- CAPAs
- Complaints
- Information sharing
FDA's documents
Content of Premarket Submissions for Management of Cybersecurity in Medical Devices
Postmarket Management of Cybersecurity in Medical Devices
Identify and resolve interoperability issues
Medical device interoperability is the ability to safely, securely, and effectively exchange and use information among one or more devices, products, technologies, or systems.
'Interoperability Concerns in Clinical Practice include
- Safety (e.g., lack of or incomplete data integration)
- Poor prioritization (e.g., alarmed devices)
- Lost and missing data
- Inefficiency (e.g., double entry of data)
- Reluctance to standardize processes
- Inability to measure care and use metrics to improve care
- Failure to transfer/disseminate successes
Interoperability challenges include:
- 'Latent (hidden) interoperability failures - failures within specific functions - that may not be obvious to the user
- Assumptions that users make about the "integrity" (the completeness of the integration) of the components
- Asynchronous evolution of interfaced or interdependent components - legacy components
- Balancing the need for standardization and innovation/customization' - AAMI-FDA Interoperability Summit
Understanding the scope of FDA's mobile apps regulation
The FDA's final guidance on mobile medical applications (apps) released on September 23, 2013, entitled 'Guidance for Industry and Food and Drug Administration Staff' informs manufacturers, distributors and other entities about how the FDA intends to apply its regulatory authorities on select mobile medical apps.
Two aspects determine the FDA's scope of regulatory oversight: (1) the functionality and intended use of the mobile app and (2) the risk posed by the app if it does not function as intended. Based on these principles, mobile medical apps are sorted into 2 groups.
Group 1: For mobile apps that may meet the definition of a medical device but pose a low risk to the public, the FDA intends to exercise enforcement discretion. The apps are divided into six functionality categories including:
- Self-management tools
- Organize & Track health information
- Education
- Tools for patients to communicate potential medical conditions
- Automate Simple Calculations for Clinical Use
- Interact with Personal Health Record (PHR) or Electronic Health Record (EHR) systems
Group 2: For mobile apps that are medical devices and whose functionality could pose a risk to the patient's safety if the app were to not function as intended, the FDA intends to apply regulatory oversight. These include
- Controlling medical devices
- Displaying, analyzing, or transmitting patient-specific medical device data
- Transforming a mobile platform into a medical device
- Interpreting patient-specific data
Getting deeper insights
Attend the seminar 'FDA's medical device software regulation strategy' to understand
- How can you anticipate and defend against the malicious remote hacking and shut down of an insulin infusion pump?
- Can one software program defeat the performance capability or back up safety features of another software program?
- When interoperability surface, which software manufacturer takes the lead to solve the problem and deal with proprietary software issues?
The seminar will focus on addressing these concerns and educating participants on FDA's recent medical device software regulation strategies.
The speaker Casper (Cap) Uldriks, brings over 32 years of experience from the FDA. He specialized in the FDA's medical device program as a field investigator, served as a senior manager in the Office of Compliance and an Associate Center Director for the Center for Devices and Radiological Health. He developed enforcement actions and participated in the implementation of new statutory requirements. His comments are candid, straightforward and of practical value. He understands how FDA thinks, how it operates and where it is headed. Based on his exceptionally broad experience and knowledge, he can synthesize FDA's domestic and international operational programs, institutional policy and thicket of legal variables into a coherent picture.




