Raw Material Compliance in a cGMP environment (Health Canada/USP/EP) - Strategy Development Considerations
Raw materials are the foundation of finished products. Even a small molecule may require about twenty or less raw materials and a large molecule biotechnology product may require as many as sixty raw materials. As such, they must meet your specifications and also the regulatory requirements. In the cGMP environment, a key to ensuring that the raw material meets your expectations and regulatory expectations is to the understand regulations, common issues, and solutions.

Drawing lessons about the regulatory expectations from past 483s and warning letters
Examining various past FDA 483s and warning letters helps in understanding the compliance expectations with regard to raw material compliance. Some of the common reasons for issuing 483s and warning letters include:
- The firm's failure to implement a system for managing quality
- Failure to define and document all quality-related activities.
- Failure to implement procedures
- Failure to have adequate written procedures for the receipt, identification, quarantine, storage, sampling, testing, handling, and approval or rejection of raw materials.
- Failure to have laboratory control records that include complete data derived from all laboratory tests conducted to ensure compliance with established specifications and standards.
- Failure to prepare adequate batch production records and record the activities at the time they are performed.
- Failure to transfer all quality or regulatory information received from the API manufacturer to customers.
- Failure of the quality unit to review batch production records prior to distribution of an API batch.
- Failure to document clean out procedures
- Failure to establish and follow a change management system evaluating all changes that could affect the production and control of your API, and failure to evaluate the potential effect changes may have on the quality of your API.
If any of the above pointers hold true with your firm, it's time to develop or reevaluate your overall strategy for raw material compliance.
Tip: To determine whether your API is in conformance with CGMP, the document considered by the FDA investigators is API CGMP Guidance.
Why should you develop a raw material compliance strategy?
Historically, only one team was involved in selecting raw materials. However, that does not work anymore. The evolving regulatory requirements, increased access to unique and complex materials, customer locations, global sources, and handling methods has necessitated expertise of various teams to be involved in selecting raw materials. Also, the raw material requirement varies across the phase1, phase 2 and phase 3. All these factors call for the development of an overall strategy. Developing an overall strategy can benefit your firm in the following ways:
- A strategy can create large savings for the firm due to appropriate testing, reusing of materials that already are being used for other products manufactured within the firm
- Can minimize the volume of testing and provide an instant history of raw materials
- Can facilitate continued use of raw materials through the 3 phases thereby minimizing testing cost
- Enable specification documents of testing and required quantity for testing
Strategy development considerations
Understand, define, specify, brainstorm
Understand: Developing an overall raw material compliance strategy requires a thorough understanding of the role of the raw materials in the manufacturing process, testing, supplier assessments as well as the regulatory requirements.
Define, Specify:Raw materials used in the process can be very diverse. The first step to developing raw material compliance strategy is to define the raw materials in consideration. The diversity of raw materials and the usage of many other raw materials in the manufacture of the final product necessitates clarity about which material definition is applied for developing the quality control strategies.
Brainstorm:On setting the definitions, have a brainstorming exercise to plan the specific elements needed for the strategy.
Apply a risk-based approach to supplier and raw materials management
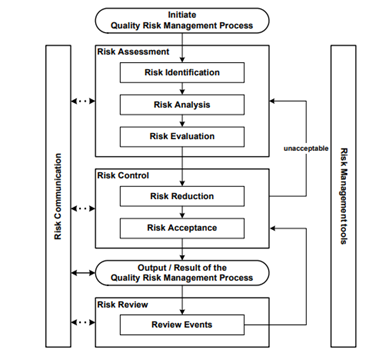
The Q9 document offers examples of tools for Quality Risk Management. It offers a systematic approach to quality risk management. The following figure from the Q9 document provides an overview of a typical risk management process:
Apply raw material testing an essential part of raw material control strategy
Following are useful resources while considering raw material testing as a strategy.
7.1 General controls
7.2 Receipt and quarantine
7.3 Sampling and Testing of Incoming Production Materials
FDA Subpart E - Control of Components and Drug Product Containers and Closures
"There shall be written procedures describing in sufficient detail the receipt, identification, storage, handling, sampling, testing and approval or rejection."
"Each lot of components, drug product, containers, and closures shall be withheld from use until the lot has been sampled, tested, or examined, as appropriate, and released for use by the Quality Control Unit"
"Representative samples of each shipment of each lot shall be collected for testing or examination. The number of containers to be sampled, and the amount of material to be taken from each container, shall be based upon appropriate criteria such as statistical criteria for component variability, confidence levels, and degree of precision desired, the past quality history of the supplier, and the quantity needed for analysis and reserve where required by 211.170"
"Samples shall be collected in accordance with the following procedures:
- (4) If it is necessary to sample a component from the top, middle, and bottom of its container, such sampling subdivisions shall not be composited for testing.
- (6) Containers from which samples have been taken shall be marked to show that samples have been removed from them."
"Samples shall be examined and tested as follows:
- (1) At least one test shall be conducted to verify the identity of each component of a drug product. Specific identity tests, if they exist, shall be used.
- (2) Each component shall be tested for conformity with all appropriate written specifications for purity, strength and quality.
- Each lot or batch of raw material shall be tested against the specifications for the raw material prior to its use in the fabrication of a drug.
- No lot or batch of raw material shall be used in the fabrication of a drug unless that lot or batch of raw material complies with the specifications for that raw material.
- Notwithstanding subsection (1), water may, prior to the completion of its tests under that subsection, be used in the fabrication of a drug.
- Where any property of a raw material is subject to change on storage, no lot or batch of that raw material shall be used in the fabrication of a drug after its storage unless the raw material is retested after an appropriate interval and complies with its specifications for that property.
- Where the specifications referred to in subsections (1), (2) and (4) are not prescribed, they shall
- a. be in writing;
- b. be acceptable to the Director who shall take into account the specifications contained in any publication mentioned in Schedule B to the Act; and
- c. be approved by the person in charge of the quality control department.'
Attend the webinar 'Raw Material Requirements (Health Canada/USP/EP) in a cGMP Environment - Issues and Solutions' to explore raw materials and their requirements - issues and solutions. It will also explore how water impacts the final product since water is the single largest raw material that is used within most processes. Another objective is to assure that your organization is maintaining itself within a cGMP compliance framework to include ICH Q7, Q9 and Q11. Case studies to include Warning Letters will be discussed to illustrate regulatory raw material issues.
The speaker Barry A. Friedman, PhD, is a Consultant in the Biotechnology, Regulatory Compliance and Aseptic Processing Arena. Dr. Friedman possesses over 30 years of industrial managerial experience in various aspects of biopharmaceuticals and medical devices to include regulatory compliance, expert witness testimony, GLP/GMP, quality control, auditing, sterility assurance, microbiological/analytical validations and fermentation technology.




