Sterility testing of Manufactured Products, and Sterility test failure Investigation Approach
Pharmaceutical and biopharmaceutical therapeutics must be sterile and safe for human use. Sterility testing is performed to ensure that the Pharmaceutical and biopharmaceutical therapeutics are actually safe. The testing method to be used for sterility testing is recommended in USP<71>. This article points out to the regulations guiding manufactured product sterility testing.

In the course of sterility testing of products, sooner or later, you will come across a sterility test failure (either real or false-positive). When faced with such a situation, many facilities do not have the Standard operating procedure (SOPs) in place to guide their further action. Even if the SOP exists, often it only provides guidance about chemistry test and not the guidance about conducting an effective, robust and compliant sterility failure investigation. The latter part of this article outlines the sterility test failure investigation approach.
Sources of contamination generally discovered during sterility testing
- Personnel
- Work surfaces
- Supplies
- Equipment
- Failure of engineering controls
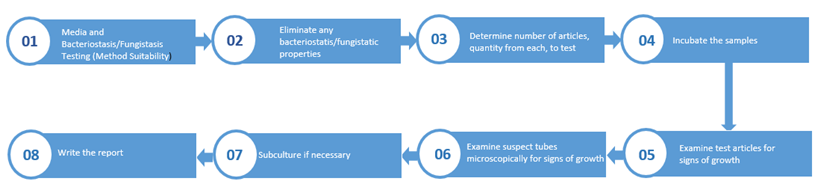
The flow of the sterility test
Regulations guiding manufactured product sterility testing
21 CFR 211.110(a) "To assure batch uniformity and integrity of drug products, written procedures shall be established and followed that describe the in-process controls, and tests, or examinations to be conducted on appropriate samples of in-process materials of each batch. Such control procedures shall be established to monitor the output and to validate the performance of those manufacturing processes that may be responsible for causing variability in the characteristics of in-process material and the drug product."
21 CFR 211.160(b) "Laboratory controls shall include the establishment of scientifically sound and appropriate specifications, standards, sampling plans, and test procedures designed to assure that components, drug product containers, closures, in-process materials, labeling, and drug products conform to appropriate standards of identity, strength, quality, and purity.
21 CFR 211.165(e) states that "The accuracy, sensitivity, specificity, and reproducibility of test methods employed by the firm shall be established and documented. Such validation and documentation may be accomplished in accordance with B' 211.194(a)(2)."
21 CFR 211.167(a) "For each batch of drug product purporting to be sterile and/or pyrogen-free, there shall be appropriate laboratory testing to determine conformance to such requirements. The test procedures shall be in writing and shall be followed."
21 CFR 211.180(e) "Written records required by this part shall be maintained so that data therein can be used for evaluating, at least annually, the quality standards of each drug product to determine the need for changes in drug product specifications or manufacturing or control procedures * * *.
21 CFR 211.192 "All drug product production and control records, including those for packaging and labeling, shall be reviewed and approved by the quality control unit to determine compliance with all established, approved written procedures before a batch is released or distributed. Any unexplained discrepancy (including a percentage of theoretical yield exceeding the maximum or minimum percentages established in master production and control records) or the failure of a batch or any of its components to meet any of its specifications shall be thoroughly investigated, whether or not the batch has already been distributed. The investigation shall extend to other batches of the same drug product and other drug products that may have been associated with the specific failure or discrepancy. A written record of the investigation shall be made and shall include the conclusions and follow up."
False-positive sterility test failure
Experienced professionals in sterility testing have found that sterility test failure investigations are sometimes flawed. - The root cause is not investigated sufficiently, the investigation summary report does not detail the investigated areas and so one cannot exactly tell the kind of investigations were performed. There is no sufficient data to support the conclusions regarding the root cause for the sterility test contamination.
Poor or deficient sterility test facilities
Is it a good practice to perform sterility tests in laminar flow hoods located in cleanroom suites to test products manufactured using advanced aseptic processing such as isolator technology? Is it a good practice to perform sterility test for products that are terminally sterilized with a qualified steam cycle? It is not for many reasons. Chances of having a false-positive sterility test are high because many cleanroom suites have air insufficient cascade to prevent microbial ingress into the testing area. Storing the sterility test samples in the micro lab until testing creates the chances for superficial contamination with viable microbes.
The right practice is to adhere to the Section XI "Sterility Testing" of the FDA's 2004 Aseptic Processing Guidance which states:
"The testing laboratory environment should employ facilities and controls comparable to those used for aseptic filling operations. Poor or deficient sterility test facilities can result in test failure (False-positive results)
If production facilities and controls are significantly better than those for sterility testing, the danger exists of mistakenly attributing a positive sterility test result to a faulty laboratory even when the product tested could have, in fact, been non-sterile. Therefore, a manufacturing deficiency may go undetected. The use of isolators for sterility testing minimizes the chance of a false positive test result."
What should the sterility failure assurance investigation approach be?
Section XI.C.1 of the FDA's 2004 Aseptic Processing Guidance describes the key elements of the investigation:
- Identification (speciation) of the organism in the sterility test
- Record of laboratory tests and deviations
- Monitoring of production area environment
- Monitoring Personnel
- Product Presterilization Bioburden
- Production record review
- Manufacturing history
Best practices:
- To perform contamination investigations, have a formal plan to follow.
- Employ a systematic approach to sterility assurance failure investigations.
- Perform investigations with an open mind to all the causes of that failure.
- Document sufficient investigation that details all the areas of the investigation.
- Have sufficient data to support the conclusions drawn regarding the root cause for the sterility test contamination.
- Perform 3 simultaneous investigations - Microbiology investigation, manufacturing investigation, validation investigation
The importance of a meaningful Environmental Monitoring EM
While reviewing historical EM data collected for a particular area is critical when a sterility test growth-positive is discovered, it is also important to discover the source of the contaminating microbe. There is a difference between the root cause and the source. The root cause tells you how the microbe got into the product but will not tell you what the source of the microbe is. Finding the source of the microbial contaminant may take hundreds of samples. Samples are taken using swabs, at non-routine sites which may not be cleaned effectively. Due diligence is required to find the source of the microbial contamination.
Attend the webinar Performing an Effective, Robust and Compliant Sterility Failure Investigation: How to Avoid Common Mistakes. It will highlight the mistakes often made when corrective and preventative actions are not clearly identified and applied during a manufactured product sterility test failure investigation because an ineffective investigational procedure and tool were used to conduct a sterility test failure investigation. The webinar will also illustrate how avoiding such common mistakes will ensure that these types of products meet the sterility requirements USP <71> and other regulatory guidelines applicable to finished products, bulk drug substance, raw materials or excipients.
The speaker Charity Ogunsanya has over 26 years of extensive experience within the Pharmaceutical, Biotechnology, Biologics, Cell-Therapy, Diagnostics, Research and Development, Radio-pharmaceutical, Contract Manufacturing Organization (CMO) and Medical Device companies.
She has been a sought-after expert and have been consistently hired after several competitive efforts by major fortune 100 companies to assume key roles specifically related to remediation and difficult Quality and Compliance related deficiencies associated with FDA's Consent Decree, FDA's Warning Letters and difficult regulatory bodies inspectional findings which is always achieved with a successful outcome.




