Understanding ISO 13485:2016 for Compliance and Quality
This article provides an overview of the major sections of ISO 13485:2016.

Why ISO 13485 is revised and approved in 2016?
Greater emphasis on risk-based approaches to quality and safety
Quality Management System
- The certification body should be in agreement as it will be stated on the ISO 13485
- Should include the list of exclusions from the standard if any
- The mandatory documented procedures required by the ISO 13485 include:
- Control of documents
- Control of records
- Internal audit
- Control of non-conforming products
- Corrective and preventive actions
- Validation of computer software
- Customer specifications (for manufacturing, inspection, packaging, and delivery)
- Monitoring and measurement
- Servicing and installation (if applicable)
- Management review
- Work environment and contamination control
- Design and development
- Validation of sterilization and sterile barrier systems (if applicable)
- Identification and traceability
- Preservation of product
- Calibration or verification for measuring equipment
- Feedback and complaint handling
- Reporting to regulatory authorities
- Advisory notices, reworks, data analysis
- Keep reference documents demonstrating conformity
- Include a description of each medical device family
- Develop and maintain procedures for each medical device family
- Develop and maintain specifications and procedures for measurement of products
- Document procedures for servicing and installation
Management Responsibility
- Complaint handling
- Reporting to regulatory authorities
- Monitoring and measurement of processes
- Monitoring and measurement of products
- Corrective action
- Preventive action
- Applicable revised regulatory requirements
- Improvement required in QMS
- Improvement of product related to customer requests
- Changes needed in response to regulatory requirements
- Resource needs
Resource Management
- Requires documented processes for establishing competence, providing training, and ensuring awareness of personnel
- Requires application of a risk-based approach for determining the methodology used to check the effectiveness of the training
Product Realization
- Established quality criteria for the product
- Defined processes and supporting documentation
- Appropriate infrastructure and work environment
- Trained and adequately qualified staff
- Established verification, validation, monitoring, measurement, inspection, handling, storage, distribution and traceability activities in line with the products and processes
- Documented records providing evidence that the product realization processes and product meet the required specifications
Measurement, Analysis and Improvement
- Feedback is not just about customer complaints. To ensure product safety and evaluate its performance, the feedback processes should be clearly defined to gather data from production and post-production activities.
- To assure that the product monitoring is being done, the feedback received should serve as input in the risk management process and product realization process.
- A new section for complaint handling and reporting to regulatory authorities is included.
- If a complaint is not investigated the justification shall be documented. Also, any corrections or corrective action as a result of the complaint received shall be properly documented.
- There is more clarity pertaining to the monitoring a measurement of processes for companies to comply with during the implementation of a quality system. These activities are conducted at applicable stages of product realization.
- There is clarity about the nonconforming product. Also there is expanded information pertaining to the handling of the nonconforming product before and after delivery to ensure that the instances are each handled appropriately.
- A new section Rework is included. The rework activities are required to be performed as per the procedures or instructions. The testing shall be in the same manner as the original product.
Many Global Regulatory bodies are including ISO 13485 into their requirements making the product suitable for companies to market internationally. The update to ISO 13485:2003 helps in aligning with the regulatory requirements with respect to Documentation, Complaint management, and reporting issues to regulators.
The ISO 13485 standard does not have a high-level Annex SL structure that's found in the other ISO 9001:2015 standards. However, it requires risk-based approaches for quality and safety protection just like the requirement that is mentioned over a dozen times in the ISO 9001:2016 standards. There is a greater emphasis on risk-based approaches to quality and safety as compared to the ISO 13485:2003.
Compliance with ISO 9001:2016 requires companies to demonstrate that risk is taken into account for the entire organization's Quality Management System (QMS) processes that include aspects shown in the following figure.
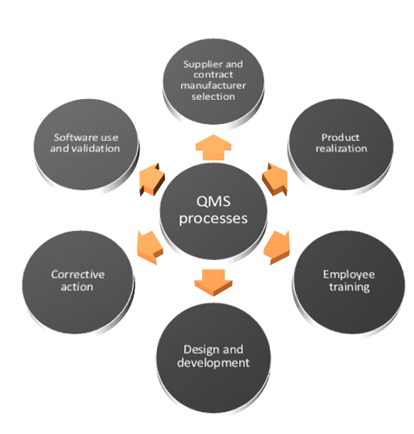
Compliance with ISO 9001:2016 requires companies to demonstrate that risk is taken into account for the entire organization's Quality Management System (QMS) processes
The following section will take you through the major sections of the ISO 13485:2016 and provide action items and best practices to help you apply them in your medical device company.
Definition: "A formal system that documents the structure, processes, roles, responsibilities and procedures required to achieve effective quality management." (ASQ, a Quality Management System).
What QMS consists of: The QMS consists of the fundamental set of policies, procedures, forms, and work instructions. It also provides the sequences, interactions and resources required to do business with a medical device company. Quality record documentation is performed to show that the QMS is executed and followed.
The regulatory requirements vary across markets. While addressing the specific regulatory requirement of the market where the medical device is to be manufactured and marketed, the QMS content should also address the requirement of the ISO 13485:2016.
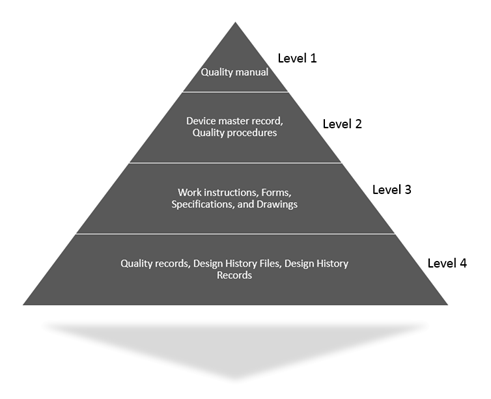
QMS Hierarchy example
Monitoring and controlling processes that impact the requirements of the risk-based approach: Whether you run the processes in-house or outsource some of them, processes that impact requirements of the risk-based approach ISO 13485:2016 must be monitored and controlled. There should be defined roles and responsibilities in documented quality agreements with any resources that are outsourced. During the continuous monitoring of the QMS for its effectiveness, adjustments should be made as necessary and documented. One of the best ways to do so is to have Key performance indicators for the processes within the QMS.
Companies can demonstrate that they are serious about protecting safety and quality by validating their computer systems. The more specific requirements for validation of systems such as Enterprise Resource Planning (ERP), QMS and Laboratory Information Management Systems (LIMS), as well as any other applications used in the development or maintenance of medical devices are in ISO 13485. As software validation consumes a lot of time and resources, many life sciences companies choose automated validation.
Even before you start using the software, it is best to customize and configure to align with ISO 13485. By establishing an automated system for validation, companies get on the fast-track for CFR part 11 compliance.
Documentation requirements
Documentation of QMS is crucial. Commit to doing the documentation right. Else, it could result in problems. Documentation of QMS processes, quality events, and workflows should be developed with great care. Documentation is about defining the processes and showing that the processes are really being followed. It provides evidence of the objective. Documentation assists your staff through design, development, manufacturing and support of medical devices. It should show evidence that the organization is committed to meeting the requirements of the QMS.
Setting up a full-fledged, functional, document management practices for your company is of the key elements of a QMS.
Quality manual
The Quality Manual is a critical part of your Quality management system. Ensure that your Quality Manual covers the following:
The Scope of the QMS:
It is a description of what your company does and the boundaries of your Quality Management system.
Documented procedures
Procedures can be shown graphically and can be included in the quality manual. If there are longer procedures requiring more written information, include references to these documents in the Quality Manual.
Descriptions of processes and their interactions: The best way to represent this is by using a top-level flowchart that illustrates the basics of the company's processes, with arrows pointing to how they interact.
Medical Device File
The requirements for the various elements of a medical device file are set forth in the Sub-clause 4.2of ISO 13485:2016.
Document Control
In adherence to Section 4.2.4 (Control of documentation) of ISO 13485:2016, documents required by the Quality Management System (QMS) should be maintained and controlled to ensure their usability, effectiveness, and adequacy for operation. In the document control procedure, define your organization's criteria for document control and ensure that the documents are reviewed and approved prior to use. Define controls to prevent unintended used of outdated documents, and track the revision status and changes of documents.
Control of records
Records should be controlled just like the way documents are. Records provide proof that the required processes have been executed.
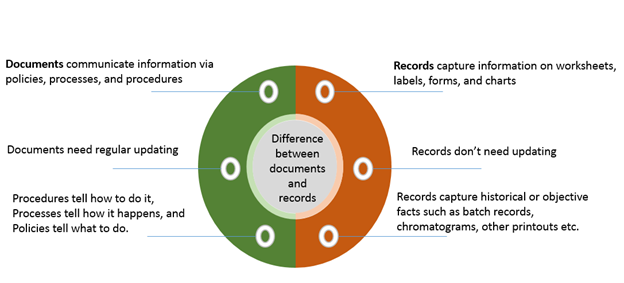
It is vital to ensure the correct understanding of what records are. Often people are a little confused between documentation and records. The difference between documents and records is illustrated below:
Even the best of control over records will fail if there is no complete support of the Executive management in maintaining product safety and promoting the ongoing improvement of processes. The executive management should do more than just pay lip service to the QMS. By embracing, supporting, and living up to it, they should foster a culture of true quality across the organization. A small slip in their approach to true quality can make it extremely difficult to restore.
The section 5 of ISO 13485:2016 specifies the requirements for management responsibility. The FDA in CFR 820.12 states, "Management with executive responsibility shall establish its policy and objectives for, and commitment to, quality. Management with executive responsibility shall ensure that the quality policy is understood, implemented, and maintained at all levels of the organization."
Section 5.6 section of ISO 13485:2016 is Management overview.
Documented procedures are required.
Inputs are expanded
The outcome should be recorded and should include the input reviewed and the decisions/actions to:

Section 6 of ISO 13485:2016 cover this topic. The Company is required to identify the need for and to allocate qualified staff, infrastructure, processes, and work environment to ensure product safety. It is important to ensure competency of staff through formal training, record management of employee competency, and providing training as necessary. Another critical component of resource management is the buildings, workspaces, process equipment, and software to support business operations, and support services.

A High-level view of what product realization includes
Product realization is a description of how your company designs, develops, manufactures, and delivers medical devices.
Product realization comprises the resources and processes needed to define customer needs, design and development, purchase, production and field assistance.
Many elements of the QMS, and a variety of staff and resources help in the outworking of product realization. In planning and expending your product realization efforts, it is critical to have the following in place:
Attend this webinar 'ISO 13485:2016 - What are the hot topics and changes?' to understand, what are the changes to the previous standard and how to implement the changes in your current quality management system in a simple and quick way.
Instructor profile: Dr. H.C. Frank Stein, medical engineer, medical engineering experience since 25 years, clinical and research experience in cardiac surgery and cardiology, industrial experience in ophthalmology, neurology, traumatology and dental implants, active implants, active devices, international project and regulatory consulting experience in Europe, North-America, Asia, Australia, Arabic Countries, Latin-America.




