Understanding Phase I and first-in-human Clinical Trials and FDA's CGMP Requirements
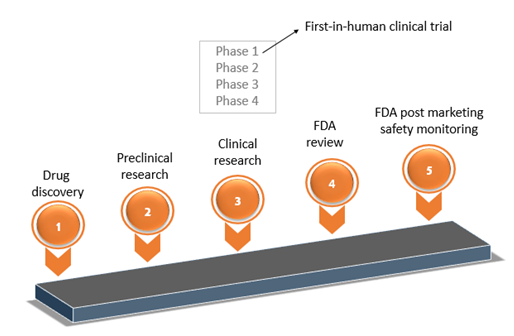
Any new drug development process involves the following steps to ensure that the product is safe and efficacious. The steps include:
- Discovery and development
- Pre-clinical research
- Clinical research
- FDA review
- FDA Post-Market Safety Monitoring

This article sheds light into phase 1 of the 3rd step which is 'first-in-human' clinical trial. Directors, managers, supervisors, lead workers in Regulatory Affairs Quality Assurance and Quality Control, workers who will prepare GMP documents for early phase products, and workers who will review GMP documents for early phase products will greatly be equipped to implement CGMP by understanding this important phase.
'The first in human studies is designed mainly to investigate the safety/tolerability (if possible, identify MTD), pharmacokinetics and pharmacodynamics of an investigational drug in humans.'
- In the pre-clinical step, the investigational medicinal product (IMP) that is developed is assessed through in-vitro or animal testing.
- The next clinical research step consists of 4 phases. In phase 1 of clinical research, the IMP is tested on human subjects for the first time.
- When an investigational medicinal product (IMP) that is developed and assessed through in-vitro or animal testing, is tested on human subjects for the first time, it is called Phase I or First-in-human/Clinical Trials.

The clinical research phase studies
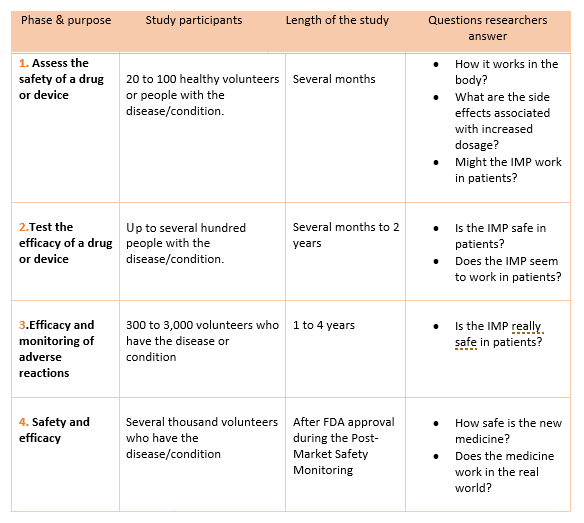
- Each phase is considered a separate trial
- Investigators are required to submit their data for approval to the FDA after completing a phase before continuing to the next phase.
- It ensures appropriate actions prior to and during manufacturing to eliminate or mitigate potential hazards to safeguard the quality of phase 1 investigational drug
Steps involved in the Investigational new drug process
- Submit IND application to FDA: Before beginning the clinical research, the sponsor or the drug developer should submit the investigational new drug (IND) application to FDA. The IND application should include animal study data and toxicity, manufacturing information, clinical protocols for studies to be conducted, data from any prior human research, and information about the investigator.
- FDA Review: The FDA which consists of a team that includes a project manager, medical officer, statistician, Pharmacologist, Pharmakineticist, Chemist, and Microbiologist will review the IND application.
- Approval, Clinical hold to delay or stop the investigation: If 'FDA is satisfied that the trial meets Federal standards, the applicant is allowed to proceed with the proposed study.' If FDA places a clinical hold, the specific reason will be provided to the applicant.

FDA's GMP requirements
In the seminar, FDA's GMP Expectations for Phase I and First-in-Man Clinical Trials, Peggy J. Berry, who is an expert in the implementation of global regulatory strategies, will help participants take a deep-dive into FDA's GMP regulations.
"Section 501(a)(2)(B) of the FD&C Act (21 U.S.C. 351 (a)(2)(B)) requires drugs, which include IND products, to comply with current good manufacturing practice as follows:
- A drug...shall be deemed adulterated...if...the methods used in, or the facilities or controls used for, its manufacture, processing, packing, or holding do not conform to or are not operated or administered in conformity with current good manufacturing practice to assure that such drug meets the requirements of this Act as to safety and has the identity and strength, and meets the quality and purity characteristics, which it purports or is represented to possess."
- An investigational drug for use in a Phase 1 study, as defined in Sec. 312.21(a) of this chapter, is subject to the statutory requirements set forth at 21 U.S.C. 351(a)(2)(B). The production of such drug is exempt from compliance with the regulations in part 211 of this chapter. However, this exemption does not apply to an investigational drug for use in a Phase 1 study once the investigational drug has been made available for use by or for the sponsor in a Phase 2 or Phase 3 study, as defined in Sec. 312.21(b) and (c) of this chapter, or the drug has been lawfully marketed. If the investigational drug has been made available in a Phase 2 or 3 study or the drug has been lawfully marketed, the drug for use in the Phase 1 study must comply with part 211 of this chapter.
FDA's GMP guidance for phase 1 clinical trial stage
FDA's guidance goals are designed to provide some clarity on the approach and expectations, help assure safe investigational products, and facilitate product development.
Manufacturers should establish manufacturing controls based on identified hazards for the manufacturing setting that follow good scientific and QC principles. These recommendations provide flexibility to the manufacturers in implementing CGMP controls appropriate to their specific situation and application."
General Requirements
- Personnel
- QC function
- Facility and equipment
- Control of components, containers, and closures
- Manufacturing and records
- Laboratory controls
- Packaging, labeling, and distributing
- Recordkeeping
For a comprehensive and systematic evaluation of the manufacturing setting (i.e., product environment, equipment, process, personnel, materials) to identify potential hazards, attend the seminar FDA's GMP Expectations for Phase I and First-in-Man Clinical Trials
- Personnel
- Qualified, experienced or trained professionals who can perform the assigned function.
- Must have relevant experience to prepare the phase 1 investigational drug and must understand QC principles and acceptable methods for complying with the statutory requirement of CGMP
- QC qualification
- Review and release components
- Review and approval of production procedures, testing procedures, and testing criteria
- Release or reject each batch upon cumulative review
- Investigate errors and initiate corrective actions
- Facility and equipment
- Sufficient space, clean environment, appropriate construction
- Appropriate lighting, ventilation, and heating
- Appropriate cooling, plumbing, washing, and sanitation
- All equipment used in the manufacture of Phase 1 drug product should be identified, well maintained, calibrated, and cleaned appropriately according to the associated written procedures
- All equipment used in the manufacture of Phase 1 drug product should be constructed with material that will not contaminate or be reactive, additive, or absorptive with the product
- Identified and documented in production records.
- Special manufacturing situations
- Multi-product facilities
- Biological/Biotechnical products
- Sterile/Aseptic processing
Attend the seminar FDA's 'GMP Expectations for Phase I and First-in-Man Clinical Trials' to understand how to take a comprehensive and systematic evaluation of the manufacturing setting. The seminar will also guide you to take appropriate actions prior to and during manufacturing to eliminate or mitigate potential hazards to safeguard the quality of the phase 1 investigational drug.
Testimonials:
- I enjoyed the practical answers and lessons learned as shared by the presenter. Grateful for the sharing of presentation material (soft copy). Thanks to ComplianceOnline for timely response and communication. Variety of choices is extensive good and easy to register.
- GMP expectations was the most valuable topic for me. Face to face interaction and networking was good.
- Instructor is very knowledgeable and good in explaining regs and guidance. Information provided along with soft copy of slide is a great idea and very helpful.
- For me the whole event was very good. I had no knowledge of the topic prior to this seminar. The presentation was excellent and the speaker was very knowledgeable and respectful.




