Understanding the FDA regulations governing advertising and promotion of drugs and medical devices
Are you responsible for implementing advertising and promotional activities in your organization?
Do you hold a key collateral role in reviewing advertising?
Are you an official related to the advertising and promotional areas?
If yes, understanding the FDA laws and regulations on regulated products governing advertising and promotion is crucial to your professional and organizational success. If your advertising and promotional efforts violate the law, it could result in colossal fines, criminal liability, including even prison time, and a huge disruption in operations while dealing with federal inquiries into legal marketing. Gaining knowledge about the FDA Boundaries for Direct-to-Consumer Advertising will bring you up to speed about when DTC advertising makes you ripe for legal picking by the FDA.

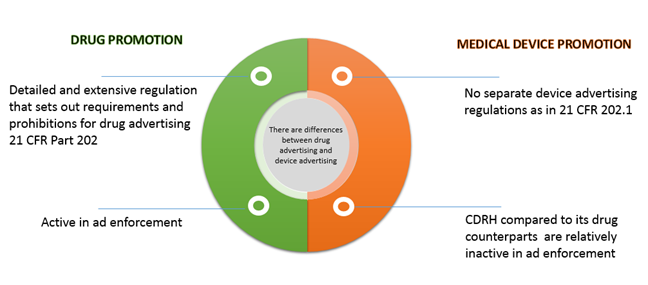
There are differences between drug advertising and device advertising. While the FDA provides detailed and extensive regulation that sets out requirements and prohibitions for drug advertising, there is the complete absence of any regulations to guide medical device companies in developing their advertising. This article provides an overview of the regulations of prescription & drug promotion and the guiding principles that govern medical device advertising and an overview of the enforcement. The relevant statutory Federal Food, Drug, and Cosmetic Act (FDCA) definitions and the FDA's drug regulatory definitions are also provided.
The Prescription and Drug promotion regulations:
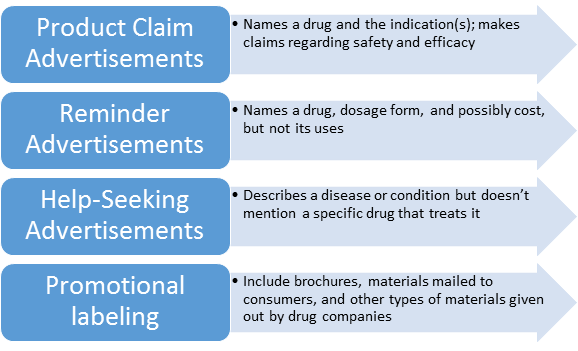
Basic types of Drug Ads:
The FDA agency requires that drug firms when providing prescription information to health professionals and consumers must be truthful, balanced, and accurately communicated. The FDA accomplishes this by means of a comprehensive surveillance, enforcement and education program.
The Prescription and Drug promotion regulations that govern prescription drug advertising and promotion for the United States are found in FDCA and Title 21 of the Code of Federal Regulations Part 202 (21 CFR Part 202)
Requirements
- Ensure that the Promote only uses that are "on label" or consistent with the FDA approved PI (Prescribing information)
- Promotional materials must not be false or misleading
- All promotional materials that include efficacy/benefit claims must provide fair balance between benefit information and information on risks associated with the product.
- Material information must be provided
The draft and final Guidance documents which provide FDA's current thinking on several topics can be accessed here.
Enforcement
The prescription drug advertising and promotion is monitored by FDA's OPDP by
- Reviewing the promotional materials submitted during the initial dissemination on Form 2253.
- Through routine surveillance
- Through the bad ad program - a program that is designed to educate health care professionals about the role they can play in helping FDA ensure that prescription drug advertising and promotion is truthful and not misleading.
If the OPDP finds violations in the promotional materials, the following types of letters can be issued identifying the corrective action:
- Notice of Violation (NOV): Known as an untitled letter, the notice of violation generally requires the company to cease the offending promotions.
- Warning letter: The warning letter can result in the company being required to do corrective advertising. It can be more laborious and costly.
Understanding the medical device advertising and promotion regulations and guiding principles
The FDA regulate the labeling of all medical devices and the advertising only of "restricted devices." (21 U.S.C. B'B' 352(a), 352(q) and (r).) - Federal Food, Drug, and Cosmetic Act of 1938, as amended (FDCA)
- Specific regulations
- 21 U.S.C. B' 352(q) provides that a restricted device is misbranded if its advertising is false and misleading in any particular, and
- 21 U.S.C. B' 352(r) provides that a restricted device is misbranded if its advertising does not contain a brief statement of the device's intended use and relevant warnings precautions, side effects and contraindications.
- Much of FDA's regulation concerning regulation of medical device promotion and advertising can be gleaned from the warning and untitled letters.
- A principle guiding acceptable medical device advertising and promotion is that any material such as print, poster boards at scientific meetings, and video must be consistent with the final product labeling of the medical device or therapy.
- Through its scientific regulations for medical devices, the FDA exerts control over advertising and promotional materials. It requires manufacturers to show that the medical device is safe and effective by using 'valid scientific evidence.' According to the FFDCA, valid evidence is
'Evidence from well-controlled investigations, partially controlled studies, studies and objective trials without matched controls, well-documented case histories conducted by qualified experts, and reports of significant human experience with a marketed device, from which it can fairly and responsibly be concluded by qualified experts that there is reasonable assurance of the safety and effectiveness of a device under its conditions of use.'
- The FDA' is sensitive to manufacturers who compare the performance of their medical technology to a competitor's product. When a competitor sees a promotional advertisement published in a medical journal and reports to the FDA's Office of Compliance, the FDA deems it as an infraction important and that it merits further investigation as seen from the numerous warning letters.
- The FDA usually applies the same kind of scientific principles it uses to assess direct claims.
- Off-label use: Off-label use is defined as "an intended use other than that in the proposed labeling. Although the FDA does not regulate the practice of medicine, it does regulate the practice of medicine when specifically promoted by the medical device industry. The FDA controls the off-label promotion of medical technology through enforcement activities.
Enforcement:
Manufacturers must take prompt action to correct the stated actions mentioned in FDA's warning letters. Failure to take prompt corrective actions can result in the FDA initiating enforcement actions including seizure, injunction, civil money penalties and criminal investigations.
Statutory definitions - Federal Food, Drug, and Cosmetic Act
Section 201(k) defines 'label' as a:
- 'display of written, printed, or graphic matter upon the immediate container of any article...'
The term 'immediate container' does not include package liners. Any word, statement, or other information appearing on the immediate container must also appear 'on the outside container or wrapper, if any there be, or the retail package of such article, or is easily legible through the outside container of wrapper.'
Section 201(m) defines 'labeling' as:
'all labels and other written, printed, or graphic matter
(1) upon any article or any of its containers or wrappers, or(2) accompanying such article' at any time while a device is held for sale after shipment or delivery for shipment in interstate commerce.
The term 'accompanying' is interpreted liberally to mean more than physical association with the product. It extends to posters, tags, pamphlets, circulars, booklets, brochures, instruction books, direction sheets, fillers, etc. 'Accompanying' also includes labeling that is brought together with the device after shipment or delivery for shipment in interstate commerce.
Drug Regulatory definitions - Advertising
21 CFR 202.1 (1)
Advertisements subject to section 502(n) of the act include advertisements in published journals, magazines, other periodicals, and newspapers, and advertisements broadcast through media such as radio, television, and telephone communication systems.
2) Brochures, booklets, mailing pieces, detailing pieces, file cards, bulletins, calendars, price lists, catalogs, house organs, letters, motion picture films, film strips, lantern slides, sound recordings, exhibits, literature, and reprints and similar pieces of printed, audio, or visual matter descriptive of a drug and references published (for example, the "Physicians Desk Reference") for use by medical practitioners, pharmacists, or nurses, containing drug information supplied by the manufacturer, packer, or distributor of the drug and which are disseminated by or on behalf of its manufacturer, packer, or distributor are hereby determined to be labeling as defined in section 201(m) of the act.
Section 201(n) states that if an article is alleged to be misbranded because the labeling or advertising is misleading, then in determining whether the labeling or advertising is misleading, there shall be taken into account (among other things) not only representations made or suggested by statement, word, design, device, or any combination thereof, but also the extent to which the labeling or advertising fails to reveal facts material in the light of such representations or material with respect to consequences which may result from the use of the article to which the labeling or advertising relates under the conditions of use prescribed in the labeling or advertising thereof or under such conditions of use as are customary or usual.
Attend the webinar 'Ensuring Compliance with Advertising and Promotional Requirements for Drugs and Medical Devices' to understand the legal and regulatory duties, as well as key recent trends in enforcement activities by the Federal Government. The course will explore in detail what FDA requires of drug and device firms as well as recent current hot buttons in FDA enforcement activity for the advertising arena.
The course Director Michael A. Swit focuses on solving the legal challenges confronted by the pharmaceutical, medical device, and other life sciences industries in tackling the myriad of legal mandates enforced by the U.S. Food & Drug Administration. Mr. Swit has extensive experience counseling life sciences firms on the demands of compliance with FDA's statutory and regulatory requirements to develop and market safe and effective drugs, biologics, medical devices, IVDs and other products. He also has advised regulated firms on a wide range of FDA regulatory matters, including drug and device approvals and marketing/promotional claims, dietary supplement health claims and regulatory issues in corporate acquisitions. His experience includes FDA development strategies, compliance and enforcement initiatives, recalls and crisis management, submissions and related traditional FDA regulatory activities, labeling and advertising, and clinical research efforts.
FAQs on FDA regulations governing advertising
1. What rules does the FDA have for advertising prescription drugs?
The FDA requires that prescription-drug advertising be truthful, not misleading, and consistent with the FDA-approved labeling.
Ads must include a “true statement” that covers side effects, contraindications, and effectiveness.
Importantly, there has to be a fair balance: promotional claims about benefits must be matched with clear, comparable descriptions of risk.
2. What is OPDP and what role does it play in drug promotion?
The Office of Prescription Drug Promotion (OPDP) is a part of the FDA that monitors drug promotion to make sure it is accurate, balanced, and legally compliant.
OPDP does this through surveillance of ads, reviewing complaints, and issuing compliance or warning letters when promotional materials violate FDA rules.
They also educate healthcare professionals through the “Bad Ad” program, which encourages reporting of misleading promotions.
3. Do companies have to submit all their drug promotional materials to the FDA?
Yes — when a company first publishes or disseminates a promotional piece (like a journal ad, sales brochure, or TV commercial), they must submit it to the FDA using Form FDA-2253.
This helps the FDA track and review how a drug is being marketed and whether it follows the approved prescribing information.
4. Can a drug manufacturer promote “off-label” uses in advertising?
No — promotion must generally stick to “on-label” uses, meaning the uses listed in the FDA-approved prescribing information.
If a company promotes a use that isn’t approved, the FDA may take enforcement action because such promotion can be considered misleading.
5. How does the FDA regulate advertising for medical devices?
The FDA regulates advertising mainly for restricted devices (those that need a prescription or special oversight).
Ads must not be false or misleading, and they must include a brief statement of intended use, plus relevant warnings, side effects, and contraindications.
Additionally, medical device promotion must be supported by “valid scientific evidence”, such as well-controlled studies, case histories, or human experience, to back up safety and effectiveness claims.
6. What happens if a company violates FDA ad rules for drugs or devices?
OPDP can issue notice of violation letters (sometimes called “untitled letters”) asking the company to stop problematic promotions.
For more serious or repeated violations, they can issue warning letters, which may require corrective advertising and more formal actions.
In extreme cases, FDA enforcement could escalate to regulatory actions such as injunctions, fines, or even criminal investigations.
7. Why does “fair balance” matter so much in drug ads?
Fair balance ensures that promotional materials don’t emphasize benefits disproportionately while downplaying risks. It’s about presenting side-effects, contraindications, and warnings in a way that’s comparable in depth to efficacy claims.
This helps patients and healthcare providers make informed decisions without being misled by overly optimistic messaging.
8. How can healthcare professionals report misleading drug ads?
Through FDA’s Bad Ad Program, healthcare professionals can report promotional materials they believe are false, misleading, or not properly balanced.
Reporting can be done via email or phone, and OPDP takes these reports seriously — it’s part of how they monitor and enforce promotional regulations




