Software Verification and Validation (V&V) Overview and must-have Documents
The regulatory bodies have specific verification and validation requirements for manufacturing and quality systems. However, there is not much understanding of the actual requirements and the best practices for meeting the requirements in the industry. The concerned personnel must understand how to meet the V& V requirements and draft a software verification and validation report package and protocol. This article presents an overview and reviews the must-have documents for V&V.

What is software verification and validation (V&V)?
Verification
820.3(a) Verification means confirmation by examination and provision of objective evidence that specified requirements have been fulfilled.
"Documented procedures, performed in the user environment, for obtaining, recording, and interpreting the results required to establish that predetermined specifications have been met" (AAMI).
Validation
820.3(z) Validation means confirmation by examination and provision of objective evidence that the particular requirements for a specific intended use can be consistently fulfilled.
- Process Validation means establishing by objective evidence that a process consistently produces a result or product meeting its predetermined specifications.
- Design Validation means establishing by objective evidence that device specifications conform with user needs and intended use(s).
"Documented procedure for obtaining, recording, and interpreting the results required to establish that a process will consistently yield product complying with predetermined specifications" (AAMI).
Validation covers 3 activities as shown below:

What are the systems that require verification and validation?
FDA requirement
Any software that is used for the design, manufacture, packaging, labeling, storage, installing and services of all finished products intended for human use should be validated.
ISO (International Organization for Standardization) and SOX (Sarbanes-Oxley) requirement
Software development process should be followed in areas where they apply to ensure that a specific process will consistently produce the expected results.
When you make an initial assessment of your system, you can arrive at a list of systems that will require validation based on the regulatory requirements mentioned above. However, there are eight common systems that fall under the control of FDA, ISO, and SOX.
- Quality management system
- Calibration management system
- Maintenance management system
- Document management/PLM system
- Quality system platforms/server validation
- Resource management system
- Clinical data tracking databases
- Labeling software
Of the above, the Document management/PLM system, Resource management system, and Quality system platforms/server validations affect multiple business units and most of these are configurable-off-the-shelf (COTS) software systems.
What are the must-have documents for software system V & V?
- Software Validation Protocol (Validation Plan): A document that outlines the project deliverables and responsibilities.
- System /Software Requirements Specification
- Network Diagram
- Risk Analysis
- 21 CFR Part 11 Compliance Analysis
- Design Specification
- Verification Protocol (Test Plan)
- Test Specifications (Test Cases)
- Requirements Traceability Matrix
- Final Validation Report
1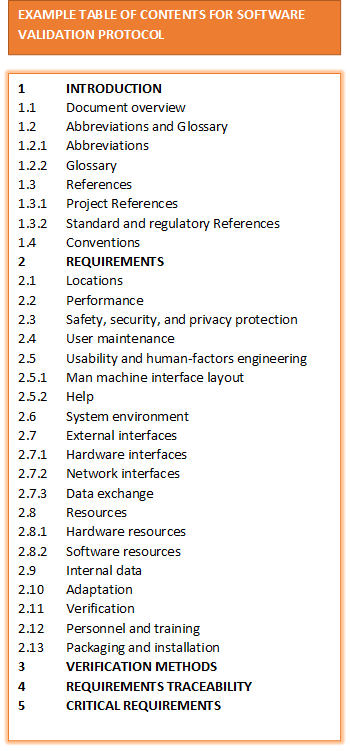 |
2 |
The network diagram document is a document required to illustrate how the system is configured on the network. It provides proof that you have a grasp of how the system is configured for your implementation.
4. Risk AnalysisThe safety of the application and identification of potential hazards is a critical requirement. The risk analysis document should evaluate safety and identify potential hazards. Rather than the traditional failure mode effect analysis (FMEA) risk assessment for the software program which are part of the device and pose a patient risk, this document should focus of the business processes that the system manages. While few risks are exclusively mitigated by the software, few risks are procedurally mitigated.
5. 21 CFR Part 11 Compliance AnalysisThe 21 CFR part 11 contains specific requirements for the use of electronic signature. The compliance analysis document evaluates the system applicability/requirements for its use as specified in the regulation.
6. Design specificationA document that indicates that the design criteria whereby the conformity with the requirement can be checked.
7. Verification Protocol (Test Plan)Contents of the verification protocol define the type of tests to be performed and the procedures and schedules for tests. Example: Acceptance criteria, materials traceability, pre and post-conditions, procedures, requirement tags, standards references.
8. Test specifications (Test cases)Test cases are documents used in the process. In software verification and validation, they are used to determine if the product is built according to the user requirements.
9. Requirements traceability matrixThis document links the requirements throughout the validation process. It ensures that all the requirements specified for a system are tested in test protocols.
10.
Attend the webinar drafting a software verification and validation report package and protocol to take a deep dive into the documentation required by the U.S. FDA for the verification and validation planning and execution of software after basic developmental testing and de-bug. In this webinar a suggested field-tested 11-element FDA model will be evaluated, implemented, with V&V documentation and test case examples. The focus of the webcast is on the most recent issues the FDA has had in this area, and remediation approaches. Software considered: 1) In-product, 2) As-product, 3) Production and test, and 4) QMS / 21 CFR Pt. 11.
The instructor John E. Lincoln has over 33 years' experience in U.S. FDA-regulated industries and 20 years as a full-time consultant. He has worked with companies from start-up to Fortune 100, in the U.S., Mexico, Canada, France, Germany, Sweden, China and Taiwan.
FAQs on Software Verification and Validation
1. What exactly is software verification vs validation (V&V)?
Verification and validation (V&V) are two complementary processes used in regulated industries to make sure software is safe, effective, and compliant. Verification is about confirming that the software was built correctly — it meets the specified design and functional requirements. Validation, on the other hand, checks whether you're building the right software — that the system fulfills its intended use in the real world.
2. Why is V&V necessary for regulated software?
Regulatory bodies like the FDA require software used in critical systems — for example, in manufacturing, labeling, or clinical data tracking — to be validated. This ensures patient safety, data integrity, and consistent performance, and it provides clear documentation to auditors and regulators.
3. Which systems typically need verification and validation under regulations like FDA, ISO, and SOX?
According to ComplianceOnline, eight common types of systems are often subject to V&V under FDA, ISO, or SOX regulation: quality management systems, calibration management, maintenance management, document management (PLM), resource management, clinical data systems, labeling software, and quality-system platforms or servers. Many of these are configurable off-the-shelf (COTS) systems, making validation especially important.
4. What are the must-have documents for a proper software V&V package?
Here are the key documents, per the article:
- Validation Plan (Protocol): Outlines project goals, scope, deliverables, and roles.
- System / Software Requirements Specification: Defines what the system needs to do.
- Network Diagram: Shows system architecture and how everything is connected.
- Risk Analysis: Identifies potential hazards and how they’re mitigated.
- 21 CFR Part 11 Compliance Analysis: Evaluates how the system handles electronic records and signatures — crucial for FDA-regulated systems.
- Design Specification: Details the design criteria that must meet the requirements.
- Verification Protocol (Test Plan): Describes types of tests, schedule, pre-conditions/post-conditions, acceptance criteria, and more.
- Test Specifications / Test Cases: Actual test cases used to verify functionality.
- Requirements Traceability Matrix (RTM): Links requirements to tests, ensuring everything is covered.
- Final Validation Report: Summarizes test results, deviations, risk mitigations, and conclusions.
5. What is risk analysis in the context of software V&V, and why is it important?
Risk analysis helps you proactively identify possible safety hazards, operational failures, or business risks in your software system. In the V&V process, this document is especially critical because it drives testing strategy, design decisions, and mitigation plans. Rather than focusing purely on system or device failure, risk analysis often centers on business processes — especially for configurable or COTS systems.
6. What role does 21 CFR Part 11 play in software V&V?
21 CFR Part 11 is an FDA regulation that governs electronic records and electronic signatures. For software used in regulated environments, a Part 11 Compliance Analysis is essential to verify whether the system meets electronic record-keeping and signature requirements, ensuring audit trails, security, integrity, and traceability.
7. How does the Requirements Traceability Matrix (RTM) help in V&V?
The RTM is a powerful document that creates a trace from each requirement (specified in the Requirements Specification) to its corresponding test case and validation status. This helps ensure nothing is missed — every requirement is verified, tested, and validated. It also provides clear evidence for audits.
8. Can you skip V&V for off-the-shelf (COTS) software?
Not always. Even if you're using COTS software, if it's part of a regulated system (e.g., quality management, or patient data management), you likely need to validate it. The article notes that many regulated systems are COTS, and proper V&V documentation is still required.
9. Why is a Final Validation Report important?
The Final Validation Report is your wrap-up: it consolidates all validation activity, test results, risk analysis, traceability, and any deviations. This is typically what auditors or regulatory bodies will examine to confirm that your system meets compliance, safety, and quality standards.
10. When should V&V planning start in the software development lifecycle?
V&V planning should ideally start very early, during or soon after system requirements definition. This ensures that validation requirements influence design, risk assessment, and test strategy from the start, making later phases smoother and more compliant.




