Issue

Mishaps in Pharmas – Causes and Clues
Access Regulatory Compliance Training sessions led by expert panelists below.
Compliance Webinars | Virtual Seminars for Professionals
Compliance Webinars | Virtual Seminars for Professionals
|
Mishaps in Pharmas – Causes and Clues |
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
The table shows problems or controversies are associated with noncompliance with GMP, inadequate clinical trials and faulty trails which resulted into producing counterfeit or faulty products. GMP Regulations Set by the United States Food and Drug Administration (FDA), GMP is a standard which stands for Good Manufacturing Practices. Importance of Following GMP Adhering to GMP regulations helps the production processes as well as the products to be effective, safe, and pure for the consumers. GMP regulations also require that the manufacturers take care to avoid contamination, errors, and mix-ups. The goal of following GMP regulations is to produce a food product or drug that the customer will want to buy and is not dangerous. Impact of Failure to comply with GMP
How to Comply with GMP
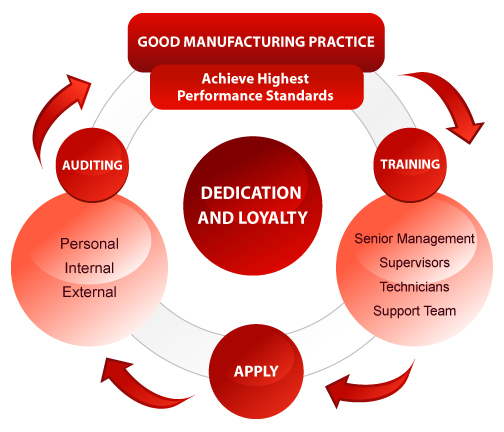
The diagram given above illustrates the approach creating and maintaining a GMP lifestyle in a company. Clinical Trails Importance of Clinical Trails Securing Safety of the Participants in Clinical Trials Sponsor
Local Site Investigator/Physician
IRBs
Regulatory Bodies
When the FDA Issue Warning Letters Key Regulations and Guidance Documents
How to Conduct Clinical Trial For
Steps for Pre-Clinical Development
According to FDA, any good clinical trial should follow the basic principles of good clinical practices (GCPs) and human subject protection (HSP). Principles of GCP are regarded as law or regulation in many countries. In FDA's regulations for conducting clinical trials, both GCP and HSP have been addressed prominently. Clinical Trial and Good Clinical Practices (GCP) Drug Labeling FDA Rules of Labeling for NDA and OTC Drugs
|
|||||||||||||||||||||||||||||||||||||||||||||
|
Drug Labeling and FDA Regulations 21 CFR PART 201—21 CFR Part 201 Subpart G --Specific Labeling Requirements for Specific Drug Products
Drug from Discovery to Marketing Reference: |
|||||||||||||||||||||||||||||||||||||||||||||
Method public java.lang.String org.ofbiz.widget.screen.ScreenRenderer.render(java.lang.String) throws org.ofbiz.base.util.GeneralException,java.io.IOException,org.xml.sax.SAXException,javax.xml.parsers.ParserConfigurationException threw an exception when invoked on org.ofbiz.widget.screen.ScreenRenderer@7d3ae094 with arguments of types [java.lang.String,]
The problematic instruction:
----------
==> ${screens.render("component://ecommerce/widget/CONew2016/BestPracticesPageScreens.xml#trainings-article-detail")} [on line 9, column 17 in component://ecommerce/webapp/ecommerce/CONew2016/bestpractices/article_detail/contentDetailMain.ftl]
----------
Java backtrace for programmers:
----------
freemarker.template.TemplateModelException: Method public java.lang.String org.ofbiz.widget.screen.ScreenRenderer.render(java.lang.String) throws org.ofbiz.base.util.GeneralException,java.io.IOException,org.xml.sax.SAXException,javax.xml.parsers.ParserConfigurationException threw an exception when invoked on org.ofbiz.widget.screen.ScreenRenderer@7d3ae094 with arguments of types [java.lang.String,]
at freemarker.ext.beans.OverloadedMethodModel.exec(OverloadedMethodModel.java:134)
at freemarker.core.MethodCall._getAsTemplateModel(MethodCall.java:93)
at freemarker.core.Expression.getAsTemplateModel(Expression.java:89)
at freemarker.core.Expression.getStringValue(Expression.java:93)
at freemarker.core.DollarVariable.accept(DollarVariable.java:76)
at freemarker.core.Environment.visit(Environment.java:221)
at freemarker.core.MixedContent.accept(MixedContent.java:92)
at freemarker.core.Environment.visit(Environment.java:221)
at freemarker.core.Environment.process(Environment.java:199)
at org.ofbiz.base.util.template.FreeMarkerWorker.renderTemplate(FreeMarkerWorker.java:257)
at org.ofbiz.widget.screen.HtmlWidget.renderHtmlTemplate(HtmlWidget.java:225)
at org.ofbiz.widget.screen.HtmlWidget$HtmlTemplate.renderWidgetString(HtmlWidget.java:270)
at org.ofbiz.widget.screen.HtmlWidget.renderWidgetString(HtmlWidget.java:130)
at org.ofbiz.widget.screen.ModelScreenWidget$PlatformSpecific.renderWidgetString(ModelScreenWidget.java:920)
at org.ofbiz.widget.screen.ModelScreenWidget.renderSubWidgetsString(ModelScreenWidget.java:104)
at org.ofbiz.widget.screen.ModelScreenWidget$Section.renderWidgetString(ModelScreenWidget.java:191)
at org.ofbiz.widget.screen.ModelScreen.renderScreenString(ModelScreen.java:396)
at org.ofbiz.widget.screen.ScreenRenderer.render(ScreenRenderer.java:135)
at org.ofbiz.widget.screen.ScreenRenderer.render(ScreenRenderer.java:97)
at sun.reflect.GeneratedMethodAccessor66.invoke(Unknown Source)
at sun.reflect.DelegatingMethodAccessorImpl.invoke(DelegatingMethodAccessorImpl.java:43)
at java.lang.reflect.Method.invoke(Method.java:498)
at freemarker.ext.beans.BeansWrapper.invokeMethod(BeansWrapper.java:866)
at freemarker.ext.beans.OverloadedMethodModel.exec(OverloadedMethodModel.java:104)
at freemarker.core.MethodCall._getAsTemplateModel(MethodCall.java:93)
at freemarker.core.Expression.getAsTemplateModel(Expression.java:89)
at freemarker.core.Expression.getStringValue(Expression.java:93)
at freemarker.core.DollarVariable.accept(DollarVariable.java:76)
at freemarker.core.Environment.visit(Environment.java:221)
at freemarker.core.MixedContent.accept(MixedContent.java:92)
at freemarker.core.Environment.visit(Environment.java:221)
at freemarker.core.Environment.process(Environment.java:199)
at org.ofbiz.base.util.template.FreeMarkerWorker.renderTemplate(FreeMarkerWorker.java:257)
at org.ofbiz.widget.screen.HtmlWidget.renderHtmlTemplate(HtmlWidget.java:225)
at org.ofbiz.widget.screen.HtmlWidget$HtmlTemplate.renderWidgetString(HtmlWidget.java:270)
at org.ofbiz.widget.screen.HtmlWidget.renderWidgetString(HtmlWidget.java:130)
at org.ofbiz.widget.screen.ModelScreenWidget$PlatformSpecific.renderWidgetString(ModelScreenWidget.java:920)
at org.ofbiz.widget.screen.ModelScreenWidget.renderSubWidgetsString(ModelScreenWidget.java:104)
at org.ofbiz.widget.screen.ModelScreenWidget$Section.renderWidgetString(ModelScreenWidget.java:191)
at org.ofbiz.widget.screen.ModelScreen.renderScreenString(ModelScreen.java:396)
at org.ofbiz.widget.screen.ScreenRenderer.render(ScreenRenderer.java:135)
at org.ofbiz.widget.screen.ScreenRenderer.render(ScreenRenderer.java:97)
at org.ofbiz.widget.screen.MacroScreenViewHandler.render(MacroScreenViewHandler.java:104)
at org.ofbiz.webapp.control.RequestHandler.renderView(RequestHandler.java:865)
at org.ofbiz.webapp.control.RequestHandler.doRequest(RequestHandler.java:582)
at org.ofbiz.webapp.control.RequestHandler.doRequest(RequestHandler.java:547)
at org.ofbiz.webapp.control.RequestHandler.doRequest(RequestHandler.java:547)
at org.ofbiz.webapp.control.ControlServlet.doGet(ControlServlet.java:224)
at javax.servlet.http.HttpServlet.service(HttpServlet.java:621)
at javax.servlet.http.HttpServlet.service(HttpServlet.java:722)
at org.apache.catalina.core.ApplicationFilterChain.internalDoFilter(ApplicationFilterChain.java:305)
at org.apache.catalina.core.ApplicationFilterChain.doFilter(ApplicationFilterChain.java:210)
at org.apache.catalina.core.ApplicationDispatcher.invoke(ApplicationDispatcher.java:749)
at org.apache.catalina.core.ApplicationDispatcher.processRequest(ApplicationDispatcher.java:487)
at org.apache.catalina.core.ApplicationDispatcher.doForward(ApplicationDispatcher.java:412)
at org.apache.catalina.core.ApplicationDispatcher.forward(ApplicationDispatcher.java:339)
at com.metricstream.SEOServlet.dispatchEcommerceRequest(SEOServlet.java:287)
at com.metricstream.SEOServlet.findPattern(SEOServlet.java:236)
at com.metricstream.SEOServlet.doGet(SEOServlet.java:49)
at javax.servlet.http.HttpServlet.service(HttpServlet.java:621)
at javax.servlet.http.HttpServlet.service(HttpServlet.java:722)
at org.apache.catalina.core.ApplicationFilterChain.internalDoFilter(ApplicationFilterChain.java:305)
at org.apache.catalina.core.ApplicationFilterChain.doFilter(ApplicationFilterChain.java:210)
at org.apache.catalina.core.StandardWrapperValve.invoke(StandardWrapperValve.java:222)
at org.apache.catalina.core.StandardContextValve.invoke(StandardContextValve.java:123)
at org.apache.catalina.authenticator.AuthenticatorBase.invoke(AuthenticatorBase.java:472)
at org.apache.catalina.core.StandardHostValve.invoke(StandardHostValve.java:168)
at org.apache.catalina.valves.ErrorReportValve.invoke(ErrorReportValve.java:99)
at org.apache.catalina.core.StandardEngineValve.invoke(StandardEngineValve.java:118)
at org.apache.catalina.valves.AccessLogValve.invoke(AccessLogValve.java:929)
at org.apache.catalina.connector.CoyoteAdapter.service(CoyoteAdapter.java:407)
at org.apache.coyote.http11.AbstractHttp11Processor.process(AbstractHttp11Processor.java:1002)
at org.apache.coyote.AbstractProtocol$AbstractConnectionHandler.process(AbstractProtocol.java:585)
at org.apache.tomcat.util.net.JIoEndpoint$SocketProcessor.run(JIoEndpoint.java:310)
at java.util.concurrent.ThreadPoolExecutor.runWorker(ThreadPoolExecutor.java:1149)
at java.util.concurrent.ThreadPoolExecutor$Worker.run(ThreadPoolExecutor.java:624)
at java.lang.Thread.run(Thread.java:748)
Caused by: org.ofbiz.widget.screen.ScreenRenderException: Error rendering screen [component://ecommerce/widget/CONew2016/BestPracticesPageScreens.xml#trainings-article-detail]: java.lang.IllegalArgumentException: Error running script at location [component://ecommerce/webapp/ecommerce/WEB-INF/actions/CONew2016/bestpractices/article_detail/trainings_articleDetail.groovy]: javax.script.ScriptException: java.lang.NullPointerException: Cannot invoke method getString() on null object (Error running script at location [component://ecommerce/webapp/ecommerce/WEB-INF/actions/CONew2016/bestpractices/article_detail/trainings_articleDetail.groovy]: javax.script.ScriptException: java.lang.NullPointerException: Cannot invoke method getString() on null object)
at org.ofbiz.widget.screen.ModelScreen.renderScreenString(ModelScreen.java:409)
at org.ofbiz.widget.screen.ScreenRenderer.render(ScreenRenderer.java:135)
at org.ofbiz.widget.screen.ScreenRenderer.render(ScreenRenderer.java:97)
at sun.reflect.GeneratedMethodAccessor66.invoke(Unknown Source)
at sun.reflect.DelegatingMethodAccessorImpl.invoke(DelegatingMethodAccessorImpl.java:43)
at java.lang.reflect.Method.invoke(Method.java:498)
at freemarker.ext.beans.BeansWrapper.invokeMethod(BeansWrapper.java:866)
at freemarker.ext.beans.OverloadedMethodModel.exec(OverloadedMethodModel.java:104)
... 76 more
Caused by: java.lang.IllegalArgumentException: Error running script at location [component://ecommerce/webapp/ecommerce/WEB-INF/actions/CONew2016/bestpractices/article_detail/trainings_articleDetail.groovy]: javax.script.ScriptException: java.lang.NullPointerException: Cannot invoke method getString() on null object
at org.ofbiz.base.util.ScriptUtil.executeScript(ScriptUtil.java:348)
at org.ofbiz.base.util.ScriptUtil.executeScript(ScriptUtil.java:324)
at org.ofbiz.widget.ModelWidgetAction$Script.runAction(ModelWidgetAction.java:416)
at org.ofbiz.widget.ModelWidgetAction.runSubActions(ModelWidgetAction.java:116)
at org.ofbiz.widget.screen.ModelScreenWidget$Section.renderWidgetString(ModelScreenWidget.java:184)
at org.ofbiz.widget.screen.ModelScreen.renderScreenString(ModelScreen.java:396)
... 83 more